Abstracts CTC-RG2017
- Abstract submission is closed
- Poster dimensions should be 48” Tall x 36” Wide
Abstracts
- John R. Shorter Male infertility is responsible for nearly half of the extinction observed in the mouse Collaborative Cross
- Daniel A. Skelly Widespread RNA splicing variation in Diversity Outbred mice
- Elizabeth G. King Genetic dissection of nutrition-induced plasticity in insulin/insulin-like growth factor signaling and median lifespan in a Drosophila multiparent population
- Rachel C. McMullan Mouse model development for exercise-induced adverse fat response
- Gregory R Keele Accounting for haplotype uncertainty in QTL mapping of multiparental populations using multiple imputations.
- Wesley Crouse Bayesian Inference of the Allelic Series at Quantitative Trait Loci in Multiparent Populations
- J. Matthew Mahoney TRiAGE: technique for ranking genes in epistasis
- Kurt H Lamour Opportunities for application of a novel targeted-sequencing technology to diverse mammalian systems
- Minako Yoshihara Design and application of a target capture sequencing of exons and conserved non-coding sequences for the rat.
- Anuj Srivastava Genomes of the Mouse Collaborative Cross
- Martin T. Ferris Sequence evolution and genetic drift across defined clusters of Mouse Inbred strains
- Abraham A Palmer Status update on the NIDA center for GWAS in outbred rats; progress, resources and opportunities.
- Leah Solberg Woods Genetic fine-mapping of visceral adiposity in outbred rats and identification of a likely causal variant in the Adcy3 gene
- Darla R Miller The Systems Genetics Core Facility at UNC
- Charles A. Phillips GrAPPA: Graph Algorithms Pipeline for Pathway Analysis
- Yangsu (Yu-yu) Ren A multi-omics approach to elucidating the genomic heterogeneity among C57BL/6 mouse substrains
- Takashi Kuramoto The forth term of the National BioResource Project-Rat in Japan
- Jennifer R Smith RGD: data and tools for precision models of human disease
- Clarissa C. Parker Genome-wide mapping of ethanol sensitivity in the Diversity Outbred mouse population
- Molly E. Harmon Particulate matter exposure effects on aryl hydrocarbon receptor activation and genetic susceptibility of respiratory disease
- Christian Fischer Genetics Browser: a new multi-dimensional web-based exploration tool
- Hemant Gujar Conserved probes in the human MethylationEPIC microarray to profile DNA methylation in mice
- Jinsong Huang Genetic modulation of bone microtraits: Joint QTL, gene expression and gene ontology (GO) analysis
- Delyth Graham Investigating a novel pathway underlying hexadecanedioate induced blood pressure elevation
- Robert W. Williams The expanded BXD family: A cohort for experimental systems genetics and precision medicine
- Spencer Mahaffey The rat based pipeline for systems genetic analysis
- Andrew B. Stiemke Systems Genetics of Optic Nerve Axon Death
- Hanifa J. Abu-Toamih Atamni Mapping novel genetic loci associated with female liver weight and fattiness variations in Collaborative Cross mice
- Hanifa J. Abu-Toamih Atamni Hepatic gene expression in health and disease using the collaborative cross mouse genetic reference population
- Hanifa J. Abu-Toamih Atamni Glucose tolerance female-specific QTL mapped in collaborative cross mice
- n Memoriam: John K. Belknap and Gerald E. McClearn, Ph.D. Founders of Quantitative Behavioral Neurogenetics and Genetics of Drug Use and Abuse
- Laurence Tecott A quantitative systems approach reveals fundamental principles of spontaneous behavioral organization exquisitely sensitive to genes and diet
- Byron C. Jones Forward genetic analysis of initial and subsequent consumption of ethanol in a large family of genetically diverse strains of mice
- Rodrigo Gularte Mérida Little evidence for transgenerational genetic effects in the trascriptome of isogenic derived mice
- Gregory W Carter Genetic variation modifies epigenetic states that mediate steroid response and gene expression QTL in mice
- David L. Aylor Genetic and environmental control of gene regulation: eQTL, epigenetics, and GxE interactions
- Allison T. Knoll Understanding heterogeneity in social behavior using QTL mapping in BXD mouse strains
- Tamara Martin Osteopontin over-expression increases H9c2 cell size
- Jeremy W. Prokop Characterization of coding and noncoding variants for SHROOM3 and chronic kidney disease from rat to human.
- Laura J Lambert Generation of new rat models of ciliopathies
- Ashley C. Johnson Identification of novel genetic factors involved in altered kidney development in the HSRA congenital solitary kidney rat
- Lauren B Shunkwiler CRISPR-Cas9 Targeting of 16q12.1 Breast Cancer Susceptibility Locus to Generate Allelic Series of Rat Mutants Results in Altered Tox3 Expression
- Isha S. Dhande A natural mutation in Stim1 creates a major defect in immune function in stroke-prone spontaneously hypertensive rats
- Olivia L Sabik Systems genetics identifies novel genes and gene networks influencing osteoblast activity
- Basel Al-Barghouthi Identification of genes affecting bone strength-related traits in Diversity Outbred mice.
- Michael-John G. Beltejar Genetic variation influences bone matrix composition resulting in varied femoral strength among inbred mice.
- Lauren J. Donoghue Identification of trans protein QTL for secreted airway mucins and a causal role for Bpifb1
- Beverly A Richards-Smith What's in a name? Standardized nomenclature for mouse and rat
- Michal Pravenec Genetic dissection of brown adipose tissue function in rat recombinant inbred strains
- Evan G Williams Metabolism in the Aging Liver: Gene-by-Environment Interactions Across the BXD Population
- Anna L. Tyler Epistatic networks jointly influence phenotypes related to metabolic disease and gene expression in Diversity Outbred mice
- Paul L Maurizio Diallel analysis reveals Mx1-dependent and independent effects driving influenza virus severity
- Sarah M. Neuner Hp1bp3 influences neuronal excitability and cognitive function
- Sumana R. Chintalapudi Cacna2d1: a novel therapeutic target for lowering IOP.
- Danny Arends Transmission distortion and genetic incompatibility of alleles in mice predisposed for obesity
- Felix L. Struebing Regulatory element networks underlying QTLs and disease loci: Towards a better understanding of non-coding variation in complex traits
- Richard S. Nowakowski Pattern detection in large datasets: comparison of methods to detect “switched” genes in gene expression datasets
- Akiko Takizawa Generation of a rat model with a one nucleotide substitution in the MAPK1 gene to mimic mutation in a patient with undiagnosed disease
- Adrianus C.M. Boon Genetic approach to study H5N1 influenza A virus pathogenesis
- David J. Samuelson Rat Mcs1b, Mcs3, and Mcs6 are genetic models of female breast cancer risk.
- Leonie C. Weber Variant alleles of estrogen receptor beta (Esr2) mediate sex dependence of traits to different extents
- Daniel M. Gatti The Effects of Sex and Diet on Physiology and Liver Gene Expression in Diversity Outbred Mice
- Danny Arends Fine mapping of a major obesity locus (jObes1)
- Pjotr Prins GeneNetwork for web-based genetic analysis
John R. Shorter1, Fanny Odet2, David L. Aylor3, Wenqi Pan2, Chia-Yu Kao5, Chen-Ping Fu 5, Andrew P. Morgan 1, Seth Greenstein5, Timothy A. Bell1,4, Alicia M. Stevans2, Ryan W. Feathers2, Sunny Patel2, Sarah E. Cates 1,4, Ginger D. Shaw 1,4, Darla R. Miller 1,4, Elissa J. Chesler6, Leonard McMillian5, Deborah A. O’Brien2,4, Fernando Pardo-Manuel de Villena1,4
1Department of Genetics, University of North Carolina, Chapel Hill, North Carolina 27599
2Department of Cell Biology and Physiology, University of North Carolina, Chapel Hill, North Carolina 27599
3Department of Biological Sciences, North Carolina State University, Raleigh, North Carolina 27695
4Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, North Carolina 27599
5Department of Computer Science, University of North Carolina, Chapel Hill, North Carolina 27599
6The Jackson Laboratory, Bar Harbor, Maine 04609
Male infertility is responsible for nearly half of the extinction observed in the mouse Collaborative Cross
The goal of the Collaborative Cross (CC) project was to generate and distribute over 1000 independent mouse recombinant inbred strains derived from eight inbred founders. With inbreeding nearly complete, we estimated the extinction rate among CC lines at a remarkable 95%, which is substantially higher than in the derivation of other mouse recombinant inbred populations. Here, we report genome-wide allele frequencies in 347 extinct CC lines. Contrary to expectations, autosomes had equal allelic contributions from the eight founders, but chromosome X had significantly lower allelic contributions from the two inbred founders with underrepresented subspecific origins (PWK/PhJ and CAST/EiJ). By comparing extinct CC lines to living CC strains, we conclude that a complex genetic architecture is driving extinction, and selection pressures are different on the autosomes and chromosome X. Male infertility played a large role in extinction as 47% of extinct lines had males that were infertile. Males from extinct lines had high variability in reproductive organ size, low sperm counts, low sperm motility, and a high rate of vacuolization of seminiferous tubules. We performed QTL mapping and identified nine genomic regions associated with male fertility and reproductive phenotypes. Many of the allelic effects in the QTL were driven by the two founders with underrepresented subspecific origins, including a QTL on chromosome X for infertility that was driven by the PWK/PhJ haplotype. We also performed the first example of cross validation using complementary CC resources to verify the effect of sperm curvilinear velocity from the PWK/PhJ haplotype on chromosome 2 in an independent population across multiple generations. While selection typically constrains the examination of reproductive traits toward the more fertile alleles, the CC extinct lines provided a unique opportunity to study the genetic architecture of fertility in a widely genetically variable population. We hypothesize that incompatibilities between alleles with different subspecific origins is a key driver of infertility. These results help clarify the factors that drove strain extinction in the CC, reveal the genetic regions associated with poor fertility in the CC, and serve as a resource to further study mammalian infertility.Daniel A. Skelly1, Narayanan Raghupathy1, Kwangbom Choi1, Anuj Srivastava1, Gary A. Churchill1
1The Jackson Laboratory, Bar Harbor, Maine 04609, USA
Widespread RNA splicing variation in Diversity Outbred mice
Alternative splicing is a ubiquitous feature of gene expression that generates a wide variety of transcript isoforms from a relatively small number of protein-coding genes. Variation in splicing is increasingly being recognized as a contributor to complex traits, including human disease. We used RNA-Seq to examine splicing patterns associated with genotype, age, tissue, and sex in Diversity Outbred mice – a multi-parent population that provides powerful and precise dissection of the genetic contribution to splicing variation. We used the RNA-Seq data to reconstruct the diploid genomes of individual mice and obtained estimates of allele-specific splice junction usage by aggregating transcript abundance across shared haplotypes. Differences in splice junction usage are widespread, and we identify numerous examples of age-, tissue-, and sex-specific splicing patterns. We mapped quantitative trait loci that drive variation in splice junction usage (sQTL). We found that sQTL are common, primarily local, and concordant across tissues. We examined conserved donor and acceptor splice site sequences but found few examples of variants that could explain the sQTL. This observation suggests that the regulation of splicing is genetically complex and may be dependent on as yet uncharacterized DNA elements. Our results highlight the power of multi-parent populations for genetic analysis of molecular phenotypes and shed new light on the role of genetic diversity as a driver of variation in transcript structure in the mouse genome.Elizabeth G. King, Patrick D. Stanley, Enoch Ng’oma, and Siri O’Day
Division of Biological Sciences, University of Missouri, 401 Tucker Hall, Columbia, Missouri 65211, USA
Genetic dissection of nutrition-induced plasticity in insulin/insulin-like growth factor signaling and median lifespan in a Drosophila multiparent population
The nutritional environments that organisms experience are inherently variable, requiring tight coordination of how resources are allocated to different functions relative to the total amount of resources available. A growing body of evidence supports the hypothesis that key endocrine pathways play a fundamental role in this coordination. In particular, the insulin/insulin-like growth factor signaling (IIS) and target of rapamycin (TOR) pathways have been implicated in nutrition-dependent changes in metabolism and nutrient allocation. However little is known about the genetic basis of standing variation in the pathways and how diet-dependent changes in IIS/TOR expression influence potentially related phenotypes. To characterize natural genetic variation in the IIS/TOR pathway, we used over 250 recombinant inbred lines (RILs) derived from a multiparental mapping population, the Drosophila Synthetic Population Resource (DSPR), to map QTL for transcript levels of the genes encoding 52 of the core components of the IIS/TOR pathway in three different nutritional environments (dietary restriction (DR), control (C), and high sugar (HS)). Nearly all genes, 87%, were significantly differentially expressed between diets, though not always in ways predicted by loss-of-function mutants. We identified cis (i.e., local) eQTL for six genes, all of which are significant in multiple nutrient environments. Further, we identified trans (i.e., distant) eQTL for two genes, specific to a single nutrient environment. A discriminant function analysis for the C and DR treatments identified which genes expression measures are most diagnostic of the diet treatment and mapping the composite discriminant function scores revealed a significant global eQTL within the DR diet. Lastly, we connect these results to a separate study assaying lifespan for 80 RILs in both DR and C diets.Rachel C. McMullan1,2, Timothy A. Bell1,2, Kunjie Hua1, Martin T. Ferris1, Vineet D. Menachery3, Ralph S. Baric3, Daniel Pomp1, Abbie Smith-Ryan1 and Fernando Pardo-Manuel de Villena1,2
1Department of Genetics, School of Medicine, University of North Carolina at Chapel Hill, NC
2Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, NC
3Department of Epidemiology, Gillings School of Global Public Health, University of North Carolina at Chapel Hill, NC
4Department of Exercise and Sports Science, College of Arts and Sciences, University of North Carolina at Chapel Hill, NC
Mouse model development for exercise-induced adverse fat response
Exercise is well known to result in beneficial health outcomes and protects against a variety of chronic diseases. Individual variation in exercise-induced responses occurs with some individuals experiencing negative responses, including fat gain. Some variation in exercise-induced responses can be attributed to exercise regimen, lack of adherence, and compensatory adaptations. However, the genetic contribution to physiological response to exercise is poorly understood. In mice, previous studies have examined physical activity levels and metabolic response. However, no systematic effort has been conducted to identify and characterize adverse responders, despite the fact that in the pre-Collaborative Cross population (partially inbred), ~17% of 176 mice gained fat in response to voluntary exercise. To address this question, we used the Collaborative Cross (CC) multi-parental population because of its high levels of genetic variation, its reproducible nature, and the observation that the CC is a rich source of novel disease models, many of which are paradoxical in nature. We examined body mass and composition response to two weeks of voluntary exercise and no exercise in multiple age-matched (~9 months) female mice from 13 CC strains. We identified CC002/Unc from this screen as an exercise-induced adverse fat responder. We observed the same exercise-induced adverse fat response in CC002/Unc females at a younger age (~4 months). In addition, we measured body composition response in four CC strains (including CC002/Unc), in both sexes and across three different types of exercise program (high intensity interval training (HIIT), moderate intensity continuous training (MICT), and no exercise (NE)). We observed a significant sex-by-exercise program interaction on fat response. Females had different fat response dependent on the type of exercise program; whereas, males had the same fat response to each exercise program. Our results provide a much needed model for studies to determine the mechanisms behind adverse metabolic responses to exercise.Gregory R Keele1, William Valdar1,2
1Department of Genetics, University of North Carolina, Chapel Hill, NC, 27599
2Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, NC, 27599
Accounting for haplotype uncertainty in QTL mapping of multiparental populations using multiple imputations.
Multiparental populations (MPP) are powerful genetic tools composed of individuals descended from a set of J known founders (generally with J > 2). These phenotypically and genetically diverse populations have been successfully used to map quantitative trait loci (QTL) underlying traits with relevance to biomedicine and agriculture. The defined linkage disequilibrium (LD) structure of MPP allows for QTL mapping through testing the association between phenotype and haplotype, rather than genetic markers as is commonly done in genome-wide association studies (GWAS). Due to the uncertainty in haplotype identity, the statistical mapping procedure used is often an approximate method that involves regression of phenotype on haplotype probabilities or dosages (ROP). This ROP approach is highly flexible, computationally efficient, and equivalenct in expectation to methods that directly model the haplotype uncertainty, such as interval mapping. However, very small probabilities/dosages of alleles can correlate strongly with phenotypes and produce spurious QTL signals, even though they most likely represent absence of the supposed strong effect allele. We propose a conservative median of multiple imputations approach to stabilize associations, and insure that results are not artifacts of the approximate regression.Wesley Crouse, Samir Kelada, William Valdar
University of North Carolina at Chapel Hill
Bayesian Inference of the Allelic Series at Quantitative Trait Loci in Multiparent Populations
Multiparent populations (MPPs) are experimental populations in which every individual’s genome is a random mosaic of a select set of known founder haplotypes. Such populations provide distinct advantages for detecting quantitative trait loci (QTL) because tests of association between phenotypes and genetic variation can leverage inferred founder haplotype descent. Once a QTL is detected, however, further analysis is required to determine how inferred haplotypes group into distinct functional alleles—the allelic series or “strain distribution pattern” (SDP). We propose a Bayesian framework for inferring the SDP that takes into account sources of uncertainty found in typical MPPs, including individuals’ haplotype state at the QTL, the number of functional alleles, and the magnitude of their effects.In applying our method to simulated and real data from incipient Collaborative Cross lines, we consider two main SDP priors, both derived from the coalescent: a default prior based on the Dirichlet process, and an informative prior based on the local or genome-wide phylogeny of the founder strains. The default prior results in low posterior certainty on any one SDP but permits high-confidence allelic contrasts and discriminatory scoring of candidate causal variants. The informative prior is powerful in identifying SDP that are consistent with phylogeny but is sensitive to misspecification of the causal region. Our method, TIMBeR, scales to MPPs with a large number of founder strains, which we demonstrate by analyzing data from the Drosophila Synthetic Population Resource, and provides a robust framework for prioritizing downstream experiments of contrasting alleles.
Kazi I Zaman1,2, Sujoy Roy2, Ramin Homayouni2,3
1Department of Computer Science, University of Memphis, Memphis, TN, 38152, USA
2Bioinformatics Program, University of Memphis, Memphis, TN, 38152, USA
3Department of Biological Sciences, University of Memphis, Memphis, TN, 38152, USA
Evaluation of gene networks using literature cohesion
BackgroundGeneNetwork [www.genenetwork.org] is a web tool that enables analysis of genetic and gene expression datasets across large panels of recombinant inbred mice [1]. Analysis of GeneNetwork data is challenging due to variability in microarray platforms, normalization methods, and biological factors. The goal of this project was to develop an analysis pipeline using literature-derived functional cohesion to evaluate GeneNetwork output and to extract meaningful insights.
Material and MethodsUsing GeneNetwork, we identified the top 200 genes whose expression levels correlated with Sirt3 expression in liver tissues across BXD recombinant inbred mice. We examined Sirt3 correlated gene networks in seven liver datasets derived from different microarray platforms and normalization methods. For two datasets, two different Sirt3 probesets were analyzed. Literature cohesion p-values (LPv) were calculated for the top 200 Sirt3 correlated genes using GeneSet Cohesion Analysis Tool [http://binf1.memphis.edu/gcat/] that was developed by our group previously [2]. To evaluate our approach, we used a gold-standard set of 429 Sirt3 target proteins, which were previously reported to be differentially acetylated in liver tissues from Sirt3 knockout mice compared with wildtype controls [3]. Recall refers to the number of overlapping genes between Sirt3-correlated gene network and the gold-standard set. Functional enrichment analysis was performed using DAVID [https://david.ncifcrf.gov/].
ResultsWe found a very high correlation (R2 = 0.97) between literature cohesion of Sirt3-correlated gene networks and recall of the gold-standard set. Functional enrichment analysis of the network with the lowest LPv revealed that the Sirt3 correlated genes belong to the following Gene Ontology classifications among many others: Mitochondrion (p-value = 4.3E-42), Oxidoreductase Activity (p-value = 2.3E-40), Lipid Metabolism (p-value = 1.2E-12), and Synthesis of Amino Acid (p-value = 1.7E-7). These results are consistent with previous reports that Sirt3 is a key regulator of mitochondrial metabolic processes [3].
ConclusionsOur results provide proof-of-concept that literature cohesion analysis can rapidly identify biologically meaningful gene networks from the vast amount of genomic data accumulating in publicly available resources such as Genenetwork.org and Gene Expression Omnibus (GEO). We posit that our approach will facilitate discovery from high throughput genomic data.
Karl W Broman
Department of Biostatistics & Medical Informatics
R/qtl2: High-dimensional data and multi-parent populations
R/qtl2 is a reimplementation of the QTL analysis software R/qtl, to better handle high-dimensional data and complex cross designs (particularly multi-parent populations such as the Collaborative Cross and Diversity Outbred mice). I'll summarize the challenges of maintaining and supporting R/qtl over the past 17 years, and will describe the features of the new software, which is split into multiple R packages. http://kbroman.org/qtl2Jane Liang, Saunak Sen
Department of Preventive Medicine, University of Tennessee Health Science Center, Memphis, Tennessee, 38163, USA
Matrix linear models for high throughput data
Genetic analysis of high-throughput phenotypes can be improved by taking advantage of known relationships between the phenotypes. Matrix linear models provide a simple framework for encoding such relationships to enhance detection of associations. Estimation of these models is challenging and computationally intensive when the datasets are large. We show that fast estimation algorithms can be developed by taking advantage of the special structure of matrix linear models. We will discuss least squares estimation including L1- and L2-penalized methods using a coordinate descent algorithm and a fast iterative shrinkage-thresholding algorithm (FISTA). Our method's performance in simulations and on an E. coli chemical genetic screen will be presented.J. Matthew Mahoney1, Anna L. Tyler2
1Department of Neurological Sciences, University of Vermont Larner College of Medicne, Burlington, VT, 05405, USA
2The Jackson Laboratory, Bar Harbor, ME, 04609, USA
TRiAGE: technique for ranking genes in epistasis
Multilocus statistical models are becoming popular in the analysis of complex traits due to their ability to identify genetic interactions, or epistasis, among alleles. These studies are limited, however, because as with single-locus trait mapping, genetic resolution of QTLs in multilocus models is typically insufficient to identify causal variants. Moreover, the combinatorial nature of multilocus models expands the pool of potential causal variants far beyond that of single-locus models. We reason, however, that epistatic interactions inherently contain more information than single-locus associations, and that this information can be harnessed to generate specific hypotheses about causal variants in interacting QTLs. In particular, a statistical interaction between QTLs implies a functional interaction between variants encoded by the interacting loci. The pool of potential causal variants in the two loci is therefore limited to those that functionally interact to influence the phenotype of interest. In recent years there has been a concerted systems biology effort to predict functional genomic networks of gene-gene interactions across species, tissues, and cell types, as well as to systematically tabulate gene-phenotype associations. Following these efforts, we developed a machine learning strategy called Technique for RAnking Genes in Epistasis (TRiAGE) that integrates functional genomic networks with epistasis to prioritize candidate gene pairs responsible for the observed epistatic interaction. TRiAGE uses gene-gene interaction weights from functional genomic networks and known gene-phenotype associations to construct a novel feature representation of phenotype-associated gene-gene interactions. TRiAGE uses these novel features to train a support vector machine (SVM) classifier to recognize known phenotype-associated gene interactions. It then classifies all putative gene-gene interactions spanning epistatic QTLs. We have performed two proof-of-concept analyses using TRiAGE to predict modifier alleles of seizure severity, first in mouse models of absence epilepsy (AE), and second in chemical induction models of seizure susceptibility (SZS) in the BXD recombinant inbred lines. In AE, TRiAGE predicts that an interaction between a known seizure gene, Plcb1, and a transcription factor regulating myelination, Tenm4, is responsible for an epistatic interaction between QTLs on Chrs 2 and 7. In SZS, TRiAGE predicts that a functional interaction between a known SZS gene, Kcnj9, and another myelin regulating transcription factor, Myt1l, is responsible for an epistatic interaction between a QTLs on Chrs 1 and 12. In both cases our predictions are highly plausible candidates, indicating that TRiAGE holds promise to nominate quality candidates for epistatic QTLs on a significantly shorter timescale than previously possible.Han Yu1, Janhavi Moharil1, Rachael Hageman Blair1
1Department of Biostatistics, University at Buffalo, Buffalo, NY, 14214, USA
BayesNetBP: An R package for probabilistic reasoning in Bayesian Networks
In this work, we present the package, Bayesian Network Belief Propagation (BayesNetBP) developed in the R programming language (https://www.r-project.org/), for probabilistic reasoning in directed networks. Network inference and analysis has become a very popular approach to understanding complex relationships and pathways from data. Bayesian Networks can be used to infer genotype-phenotype networks. The structure of these networks can guide in the prioritization of candidate biomarkers and generate hypothesis that may motivate future experiments and provide a better understanding of a disease. Although the structure is useful, it is a pre-mature endpoint. Bayesian Networks can be used for probabilistic reasoning, which adds an invaluable layer to the graphical models. Probabilistic reasoning allows an investigator to fix variables (nodes) at certain levels, akin to a perturbation, and predict probabilistic quantities of other nodes in the network (system-wide changes).
The BayesNetBP package has several major advantages. Specifically, the package (1) is the first to facilitate probabilistic reasoning in discrete, continuous and mixed BNs, (2) is the first to enable probabilistic reasoning in Conditional Gaussian Bayesian Networks (CG-BNs), which currently requires commercial software, (3) provides novel systems-level visualizations for probabilistic reasoning in the network, (4) connects seamlessly with existing graphical modeling tools in R, and (5) is also supported through an Shiny app that is accessible to the non-technical expert. Genotype-phenotype networks motivate our examples, and we present several examples using expression QTL data in discrete and mixed Bayesian Networks. However, probabilistic reasoning is facilitated through the process of belief propagation and is widely applicable to any Bayesian Network. BayesNetBP fills a major gap in the graphical modeling tools available in R.
Beth L. Dumont
The Jackson Laboratory, 600 Main Street, Bar Harbor, Maine 04609.
Variation and Genetic Control of Mutation Rates in House Mice
Mutation provides the ultimate source of all new alleles in populations, including variants that drive evolutionary adaptation and cause disease. At the same time, the de novo mutation rate is itself a quantitative genetic trait that displays striking differences between species and among individuals. Despite the central significance of this variation for genetics and evolution, little is known about the genetic causes of mutation rate heterogeneity or inter-individual variation in mutation rate. Toward these goals, I am conducting two parallel bioinformatic analyses of whole genome sequences from house mice. First, I am utilizing the Collaborative Cross (CC) 8-way recombinant inbred mouse panel as a forward evolution resource to study the accumulation of mutations over ~30 generations of organized outcrossing and inbreeding. I show that the number of accumulated mutations in the genomes of different CC lines varies 3.5-fold. This variation is driven, in large part, by the unique, dynamic genome captured in each CC breeding funnel. Building on this recognition, I perform a genome-wide scan for mutation rate modifiers and identify multiple putative mutator alleles that function in DNA repair and the cellular metabolism of genotoxic compounds. Second, I am mining high-quality genome sequences from 69 inbred laboratory strains to identify variants that are private to individual strains. These private alleles reflect recent germline mutation events that collectively mirror the action of mutational processes at work on a specific genetic background. I use this insight to show that the allelic spectrum of inherited mutations is variable among strains, with closely related strains sharing similar profiles. These findings reveal a genetic component to the nucleotide distribution of de novo mutations in the mouse genome. Together, these on-going investigations have unveiled marked complexity in germline mutation rate variation, with multiple genetic factors shaping both mutation frequency and spectrum.Kurt H Lamour1
1Department of Entomology and Plant Pathology, University of Tennessee, Knoxville, USA
Opportunities for application of a novel targeted-sequencing technology to diverse mammalian systems
In the post-genomic age, a key challenge is securing sufficient resources to assess known diversity for large numbers of samples. Dr. Kurt Lamour, a professor and molecular epidemiologist at the University of Tennessee, has developed a multiplex PCR-based pre-sequencing technology suitable for low cost, high capacity targeted sequencing. It is being used to genotype diverse organisms (microbes, plants, insects and animals) and allows rapid assessment of 100’s or 1000’s of target regions in 100’s to 1000’s of individual samples – even regions exhibiting exceptional diversity (Figure 1). More recently, the technology is being used for gene profiling of archived human tumor tissues (Targeted RNA-SEQ). Here the multiplexed gene targets are amplified with 3 to 5 housekeeping genes and the sequence coverage normalized to the ‘within tube’ housekeepers – providing unprecedented accuracy and dynamic range; at a fraction of the cost of qPCR or custom array technologies. Dr. Lamour is looking to test the technology on a wider array of organisms and scenarios and this talk will briefly overview recent results and provide an opportunity for discussion of potential applications and collaborations. The technology requires relatively little template (DNA or cDNA) and works well with mixed samples (e.g. DNA extracted from infected tissues to profile specific pathogen or host regions).
Figure 1. One locus of a 200-target multiplex used to study a diploid species of plant. Note the complex SNP and INDEL polymorphisms and the two distinct haplotypes (H1 and H2).

Minako Yoshihara1, Osamu Ohara2,3, Takashi Kuramoto4, Mikita Suyama1
1Division of Bioinformatics, Medical Institute of Bioregulation Bioregulation, Kyuhsu University
2Laboratory for Integrative Genomics, RIKEN Center for Integrative Medical Sciences
3Department of Technology Development, Kazusa DNA Research Institute
4Institute of Laboratory Animals, Kyoto University
Design and application of a target capture sequencing of exons and conserved non-coding sequences for the rat.
Since the first report on the identification of causative mutation for a monogenic disease in 2010, exome analysis has been successfully applied to more than 100 mendelian disorders. Currently, several commercial kits for exome capture are available for human and some model organisms such as mouse, bovine, and zebrafish. Although rat is widely used as an animal model for human diseases for a long time, an exome capture kit has not been designed so far. In this study, we designed a probe set for rat expanded exome sequencing, TargetEC (target capture for exons and conserved non-coding sequences), which covers not only for exonic region but also conserved non-cording sequences (CNSs) among mammalian species [1]. These conserved sequences might contain cis-regulatory elements for gene expression, thus making it possible to identify regulatory mutations. We analyzed the target capture sequencing data to identify a novel causative mutation of white spots on the head of KFRS4/Kyo. We successfully identified a deletion of approximately 50 kb in length approximately 50 kb upstream of Ednrb (Figure 1) [2]. A comparative analysis with the epigenomic data in the corresponding region in humans and mice showed that one of the deleted CNSs might be an enhancer. Further comparison with Hi-C data, which provide information about chromosome conformation, indicated that the putative enhancer is spatially close to the promoter of Ednrb, suggesting that it acts as an enhancer of Ednrb.
Figure 1. A deletion found upstream of Ednrb in KFRS4/Kyo. The genomic intervals of chr15:91,470,000-91,670,000 is shown.
George W Nelson1, Carl McIntosh2, James Lautenberger1, Sandra Burkett3, and Alan O Perantoni4
1Center for Cancer Research (CCR) Bioinformatics Resource, Frederick National Laboratory, Frederick, MD 21702
2Carl McIntosh, Genomics Analysis Unit, CCR, NCI, Frederick, MD 21702
3Molecular Cytogenetics Core Facility (MCGP), CCR, NCI, Frederick, MD 21702
4Cancer and Developmental Biology Laboratory, CCR, NCI, Frederick, MD 21702
Sequencing 71 backcross rats reveals mismaps in the RN6 reference sequence
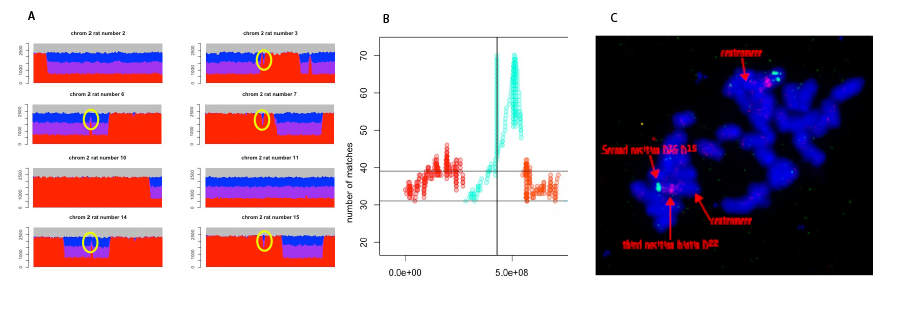
The Noble rat is susceptible to a nephroblastoma analogous to a human Wilms tumor if exposed to the carcinogen N-ethylnitrosourea in utero.(1,2) To map a locus for this susceptibility, we generated 71 rats of Noble/Fischer hybrid backcrossed to Fischer, which lacks the susceptibility. As a commercial rat genome chip was not available, we sequenced the rats at very low (~2x) coverage, reasoning that since most SNP differences between these inbred strains are fixed, calling a small fraction of all SNPs would determine recombination points. This sequencing clearly differentiated Fischer homozygous regions from Noble-Fischer heterozygous regions (Fig. 1A). However, there were 25-30 small sharply defined regions across the genome, within which the genome parentage (homozygous Fischer or heterozygous Noble-Fischer) had no correlation with that of the surrounding region. The boundaries of these regions were extremely consistent between different rats (Fig. 1A). Clear boundaries could be defined for 20 of these regions. The most plausible explanation was that these regions were mismapped in the reference genomes, both RN5 and RN6. We used a standard mapping strategy of counting matches of parentage assignment in these regions with parentage assignment across the genome, for all rats (Fig. 1B). For several of these regions there was extremely strong statistical support for a new mapped location. We are using fluorescence in situ hybridization (FISH) to confirm the alternative mapping. Figure 1C shows FISH with probes chosen for RN6 sequence within the anomalous chr. 2 region (green) and probes for sequence flanking the same region (red). If the RN6 assembly were correct, the probes should be colocalized. However, while the red probes appear close to the center of the chromosome, consistent with RN6, the green probes appear distal, close to the end of the chromosome, inconsistent with RN6 but consistent with our reassignment. The RN6 assembly has 267 unmapped fragments carrying informative SNPs, comprising ~78 Mbp of sequence. We have so far been able to define locations—within the average recombination distance of 2 Mbp of our data—for 80 of these fragments. We anticipate that our sequencing results will lead to substantial improvement in the accuracy and completeness of the rat reference genome.
Figure 1. Chromosome 2 anomalous region. A) Smoothed sequencing calls for SNPs with fixed differences between Noble and Fischer, colored by alleles seen: Blue—only Noble; red—only Fischer, purple—both. Blue-purple-red regions are heterozygous Noble/Fischer, but because of low sequencing density only one allele is seen for many SNPs. Regions inconsistent with surrounding, repeated between rats, are circled. B) Count of matches of parentage calls for this region across chr 1-3. Vertical line indicates alignment position; by identity 71 of 71 rats are consistent here, but there is no continuity with the surrounding region. 71 of 71 rats are also consistent at a position at a position much closer to the distal end of chr 2. C) FISH with probes (green) for sequence within the anomalous region and (red) for sequence in flanking regions, indicating the correct mapping of this region is at the region closer to the chromosome end.
Anuj Srivastava, Andrew P. Morgan, Maya L. Najarian, Vishal Kumar Sarsani, J. Sebastian Sigmon, John R. Shorter, Paola Guisti-Rodríguez, Martin T. Ferris, Patrick Sullivan, Pablo Hock, Darla R. Miller Timothy A. Bell, Leonard McMillan, Gary A. Churchill, and Fernando Pardo-Manuel de Villena
The Jackson Laboratory, Bar Harbor, Maine 04609, and Department of Genetics, Lineberger Comprehensive Cancer Center, Curriculum of Bioinformatics and Computational Biology, Department of Computer Science, and Curriculum of Genetics and Molecular Biology, University of North Carolina, Chapel Hill, North Carolina 27599
Genomes of the Mouse Collaborative Cross
The Collaborative Cross (CC) is a multiparent panel of recombinant inbred (RI) mouse strains derived from eight founder laboratory strains. RI panels are popular because of their long-term genetic stability, which enhances reproducibility and integration of data collected across time and conditions. Characterization of their genomes can be a community effort, reducing the burden on individual users. Here we present the genomes of the CC strains using two complementary approaches as a resource to improve power and interpretation of genetic experiments. Our study also provides a cautionary tale regarding the limitations imposed by such basic biological processes as mutation and selection. A distinct advantage of inbred panels is that genotyping only needs to be performed on the panel, not on each individual mouse. The initial CC genome data were haplotype reconstructions based on dense genotyping of the most recent common ancestors (MRCAs) of each strain followed by imputation from the genome sequence of the corresponding founder inbred strain. The MRCA resource captured segregating regions in strains that were not fully inbred, but it had limited resolution in the transition regions between founder haplotypes, and there was uncertainty about founder assignment in regions of limited diversity. Here we report the whole genome sequence of 69 CC strains generated by paired-end short reads at 303 coverage of a single male per strain. Sequencing leads to a substantial improvement in the fine structure and completeness of the genomes of the CC. Both MRCAs and sequenced samples show a significant reduction in the genome-wide haplotype frequencies from two wild-derived strains, CAST/EiJ and PWK/PhJ. In addition, analysis of the evolution of the patterns of heterozygosity indicates that selection against three wild-derived founder strains played a significant role in shaping the genomes of the CC. The sequencing resource provides the first description of tens of thousands of new genetic variants introduced by mutation and drift in the CC genomes. We estimate that new SNP mutations are accumulating in each CC strain at a rate of 2.4 6 0.4 per gigabase per generation. The fixation of new mutations by genetic drift has introduced thousands of new variants into the CC strains. The majority of these mutations are novel compared to currently sequenced laboratory stocks and wild mice, and some are predicted to alter gene function. Approximately one-third of the CC inbred strains have acquired large deletions (.10 kb) many of which overlap known coding genes and functional elements. The sequence of these mice is a critical resource to CC users, increases threefold the number of mouse inbred strain genomes available publicly, and provides insight into the effect of mutation and drift on common resources.Martin T. Ferris1, Colton Linnertz1, John Sebastian Sigmon2, Maya Najarian2, Anwica Kashfeen2, Andrew P. Morgan1, Timothy A. Bell1, Leonard McMillan2, Fernando Pardo-Manuel de Villena1
1Department of Genetics and
2Department of Computer Science, University of North Carolina at Chapel Hill
Sequence evolution and genetic drift across defined clusters of Mouse Inbred strains
Inbred mouse strains have long proven an invaluable resrouce for biomedical research, most notably for their stable and reproducible genomes. Recent studies have highlighted the phenotypic and medical importance of genetic variants that arise due to genetic drift within closely related mouse strains. Here, we report on new whole-genome sequences from a set of 31 inbred mouse strains from 9 groups of strains in broad usage across the research community. Each of these 9 strain clusters contains a set of substrains whose genomes differ only by genetic drift following propagation of independent lineages each derived from a single inbred strain. We will discuss the extent to which variants have arisen between ‘nearest neighbors’ on the phylogenetic trees within each of these clusters, and also discuss the overall characteristics of those mutations that have arisen across these stocks.Abraham A Palmer1
1University of California San Diego; Department of Psychiatry
Status update on the NIDA center for GWAS in outbred rats; progress, resources and opportunities.
The NIDA center for GWAS in outbred rats is using N/NIH heterogeneous stock rats to map a variety of drug-abuse relevant traits. N/NIH rats are the product of an 71-way cross of inbred rat strains and are thus conceptually similar to various 71-way mouse intercrosses (e.g. HS, DO). The HS rat colony is maintained by Leah Solberg Woods at Wake Forest University. Phenotyping is being performed for Pavlovian conditioned approach by Terry Robinson and Shelly Flagel at The University of Michigan; for behavioral regulation, delay discounting, sustained attention by Jerry Richards and Paul Meyer at SUNY Buffalo); and for iv nicotine self-administration by Hao Chen at University of Tennessee Health Sciences Center. Additional funded projects that build on the center’s core expertise include studies of iv cocaine self-administration by Olivier George at The Scripps Research Institute (U01), studies of bone dynamics by Douglas Adams and Cheryl Ackert-Bicknell at University of Connecticut and SUNY Rochester (R01) and studies of microbiome, metabolome and epigenome by Abraham Palmer, Mo Jain, Francesca Telese and Amalie Baud at the University of California San Diego and numerous collaborators who are receiving tissue samples that allow them to study physiological traits. We have developed an innovative genotyping-by-sequencing approach that, in conjunction with imputation to reference genomes, allows us to obtain millions of SNP markers per individual. We are performing RNAseq to obtain gene expression data for expression QTLs (eQTL) mapping. All data are stored in a sophisticated database to enable collaborations. We are in the process of transferring these data to genenetwork.org. There are numerous opportunities for collaboration that we are eager to explore; please visit www.ratgenes.org.Support: this work is supported by P50DA0377144, U01DA043799 and R01AR0707179.
Leah C Solberg Woods4, Gregory R. Keele1, Jeremy W. Prokop2, Hong He3, Katie Holl3, John Littrell3, Aaron Deal4, Sanja Francic5, Leilei Cui5, Maie Zagloul1, Yuying Xie1, Brittany Baur3, Joseph Fox3, Melanie Robinson2, Shawn Levy2, Richard Mott5, William Valdar1
1University of North Carolina, Department of Genetics, Chapel Hill, NC
2Hudson Alpha Institute, Huntsville, AL
3Medical College of Wisconsin, Department of Pediatrics, Milwaukee, WI
4Wake Forest School of Medicine, Department of Internal Medicine, Winston Salem, NC
5University College London Genetics Institute, London, UK
Genetic fine-mapping of visceral adiposity in outbred rats and identification of a likely causal variant in the Adcy3 gene
Obesity and overweight are major risk factors for multiple diseases. Although human genome-wide association studies have identified several genes for adiposity traits, these genes explain a small percentage of the heritable variance. The goals of the current study are to fine-map adiposity traits and identify underlying candidate genes using an outbred rat model: heterogeneous stock (HS) rats. We measured adiposity traits, including body weight and visceral fat pad weight, in 792 adult male HS rats. Rats were genotyped using a 10K single nucleotide polymorphism array. Quantitative trait loci (QTL) were identified using a mixed model that accounts for the complex family structure of the HS. Statistical approaches and protein modeling were used to identify underlying candidate genes and variants. We detected a 6.15 Mb QTL for retroperitoneal fat pad weight on rat chromosome 6 and demonstrate that the WKY founder haplotype at this locus results in lower fat pad weight. We identified a non-synonymous variant at a highly conserved site within the Adcy3 gene private to the WKY founder strain. Protein modeling of this amino acid change demonstrates that the leucine to proline change results in a bend in the helix which may alter membrane interactions and binding, suggesting a function role of this variant. We also identified a 3.35 Mb QTL for body weight on rat chromosome 4, although none of the genes in this region stand out as obvious candidates. These data demonstrate the power of outbred HS rats for genetic fine-mapping of body weight and visceral adiposity and implicate a variant with the Adcy3 gene as playing a role in visceral adiposity.Darla R Miller1,2, Sarah E Cates1, Kayla Harrison1, Pablo Hock1,2, Chia-Yu Kao4, Kenneth F Manly1, Ginger D Shaw1,2, Britannia Wanstrath1, Leonard McMillan4, Fernando Pardo-Manuel de Villena1,2
1Department of Genetics, UNC at Chapel Hill, Chapel Hill, NC
2Lineberger Comprehensive Cancer Center, UNC at Chapel Hill, Chapel Hill, NC
3Department of Computer Science, UNC at Chapel Hill, Chapel Hill, NC
The Systems Genetics Core Facility at UNC
The Systems Genetics Core Facility at UNC provides Collaborative Cross mice; genotyping services using the MUGA arrays and tools to utilize both the mice and the genotypes.
The Collaborative Cross is a genetic reference population derived from eight inbred strains by an international consortium of researchers. The Systems Genetics Core Facility (SGCF; http://csbio.unc.edu/CCstatus/index.py?run=AvailableLines) at UNC distributes CC lines that have reached a defined minimum level of inbreeding. The SGCF has also reconstructed the founder mosaic of each CC line, both from Most Recent Common Ancestors (MRCAs) as well as from one sequenced male from each strain.
The SGCF has distributed CC mice to >40 laboratories. CC projects fall into four categories: Strain surveys to determine the genetics of a wide variety of traits; Follow up experiments in smaller sets of CC strains; Development of models of human disease and Identification and genetic and molecular dissection of novel biological phenomena. Manuscripts using the CC have been published including a strain survey for susceptibility of Ebola virus infection, a new mouse model for spontaneous colitis, the discovery of a meiotic drive system and a new parent of origin effect on gene expression. We will present summaries on the status of the CC population (number of lines, inbreeding and breeding performance); use of the CC and publications.
Genotyping arrays using iterations of the Mouse Universal Genotyping Array (MUGA) have been designed at UNC and implemented through GeneSeek (Neogen) in Nebraska. MUGA (7000 SNPs), MegaMUGA (70,000 SNPs) and GigaMUGA (150,000 SNPs) have each required separate and improved tools which are all available at http://csbio.unc.edu/CCstatus/index.py?run.
Cai, Yanwei; Oreper, Daniel; Tarantino, Lisa M; Pardo-Manuel de Villena, Fernando; Valdar, William
Inbred Strain Variant Database (ISVdb): A repository for probabilistically informed sequence differences among the Collaborative Cross strains and their founders.
The Collaborative Cross (CC) is a panel of recently established multiparental recombinant inbred mouse strains. For the CC, as for any multiparent population, effective experimental design and analysis benefits from detailed knowledge of the genetic differences between strains. Such differences can be directly determined by sequencing; but until now whole genome sequencing was not publicly available for individual CC strains. An alternative, and complementary approach, is to infer genetic differences by combining two pieces of information: probabilistic estimates of the CC haplotype mosaic from a custom genotyping array, and probabilistic variant calls from sequencing of the CC founders. The computation for this inference, especially when performed genome-wide, can be intricate and time-consuming, requiring the researcher to generate non-trivial and potentially error-prone scripts. To provide standardized, easy-to-access CC sequence information, we have developed the Inbred Strain Variant Database (ISVdb). The ISVdb provides, for all the exonic variants from the Sanger Institute mouse sequencing dataset, direct sequence information for CC founders, and critically, the imputed sequence information for CC strains. Notably, the ISVdb also: 1) Provides predicted variant consequence metadata; 2) Allows rapid simulation of F1 populations; 3) Preserves imputation uncertainty, which will allow imputed data to be refined in the future as additional sequencing and genotyping data is collected. The ISVdb information is housed in an SQL database and is easily accessible through a custom online interface.Charles A. Phillips 1, Yuping Lu1, Erich J. Baker2, Elissa J. Chesler3, Michael A. Langston1
1Department of Electrical Engineering and Computer Science, University of Tennessee, Knoxville, TN 37996
2Computer Science Department, Baylor University, Waco, TX 76798
3The Jackson Laboratory, Bar Harbor, ME 04609
GrAPPA: Graph Algorithms Pipeline for Pathway Analysis
The Graph Algorithms Pipeline for Pathway Analysis (GrAPPA) is a web-based system designed to streamline the graph-theoretical analysis of large-scale biological data. GrAPPA is built upon Galaxy, a well-established open source platform for computational data analysis. It provides easy customization, modular extensibility, and ongoing support and development. A seamless interface permits access to numerous computational tools, including graph creation through a variety of statistical similarity metrics, graph decomposition, dense subgraph extraction and machine learning. With just a few clicks, the user can upload raw data, select from an assortment of algorithmic tools, view the results, and link to other platforms such as GeneNetwork and GeneWeaver. A built-in history function stores the results of previous analyses. An integrated help function provides on-the-fly support.Yangsu (Yu-yu) Ren, Abraham Palmer
Department of Psychiatry, University of California San Diego, San Diego, CA
A multi-omics approach to elucidating the genomic heterogeneity among C57BL/6 mouse substrains
The C57BL/6 (B6) mouse is the most widely used inbred mouse strain for laboratory studies, yet many researchers are agnostic to the genomic heterogeneity among the vast number of B6 substrains, which can drastically confound experimental results and replication efforts. In addition, despite the increasing number of phenotypic studies showing B6 substrain differences, our understanding of the causal genomic elements remain extremely limited. My goal is to identify and catalog mutations, structural variations, and expression quantitative trait loci (eQTL) that drive between-substrain differences using a panel of B6 substrains. All substrains used are derived from the same ancestral inbred strain, and their relationships to one another are supported by extensive historical records. Given that these substrains are derived from a single inbred progenitor, I expect them to be nearly-isogenic with all variation attributable to recent de novo mutations. From whole-genome sequence (WGS) data we generated at an average of >30X coverage, I found that the genomic relatedness matches the documented records of ancestry and strain derivation. In addition, I identified many interesting identify structural variations (SV) that are unique to one or more of the substrains, including insertions and deletions (Indels) and copy number variants (CNVs). Furthermore, we performed RNA-sequencing using hippocampus tissue from all of the substrains, and found that the sustrain distribution patterns of gene expression differences are consistent with those of the DNA mutations. I expect the observed differential expression to reflect the effects of de novo mutations, and by assuming that the gene expression differences are due to nearby mutations, I identified numerous cis-acting eQTLs that are statistically significant across the genome. My findings will be valuable for any current and future studies using B6 substrains.Takashi Kuramoto, Miyuu Tanaka, Birger Voigt, Yuki Neoda, Zonghu Cui, Kazumi Hagiwara, Yuki Nakagawa, Tomomi Nagao, Satoshi Nakanishi, Ken-ichi Yamasaki, Masahide Asano
Institute of Laboratory Animals, Graduate School of Medicine, Kyoto University, Yoshidakonoe-cho, Sakyo-ku, Kyoto, Japan
The forth term of the National BioResource Project-Rat in Japan
National BioResource Project-Rat (NBRP-Rat) is now operating in its 16th year and contributes as the largest rat repository to various fields of biomedical research. The major goal of NBRP-Rat comprises the collection of rat strains, the cryopreservation of embryos and sperm, and the worldwide supply of these rat strains. By March 2017, 837 rat strains are deposited to the NBRP-Rat, of which 94 strains are kept as live animals, 438 have already been preserved as embryos and 370 are stored as frozen spermatozoa. Rat strain information, Phenome data, Phylogenetic tree, SSLP and SNPs, Reproductive technologies, Whole genome sequencing of F344/Stm rat strain, GFP rats, Functional polymorphisms, BAC browser, Rat mutant map, RI strain information, and ENU mutant archives, are now available from our NBRP-Rat website at http://www.anim.med.kyoto-u.ac.jp/nbr. Recent progressing technologies for genetically modified rats (GMR); zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and CRISPR/Cas can provide thousands of useful rat models for functional genomics and human diseases.Hao Chen1, Victor Guryev2, Megan K. Mulligan3, Eva Redei4, Robert W. Williams3
1Department of Pharmacology, University of Tennessee Health Science Center, Memphis, TN, USA
2European Research Institute for the Biology of Ageing, University of Groningen, University Medical Center Groningen, Groningen 9713AD, The Netherlands
3Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Center, Memphis, TN, USA
4Department of Psychiatry and Behavioral Sciences, Feinberg School of Medicine, Northwestern University Chicago, IL, USA
Sequence variation between a genetic rat model of depression and its control strain
Major depressive disorder (MDD) is a leading cause of disability worldwide. Heritability of MDD is estimated to be between 28–44%, although the causal DNA variants underlying depression remain elusive. The WKY rat strain is a well-established model of depression. Dr. Redei obtained nearly inbred WKY stock from Harlan Laboratories in the mid-1990s and selectively bred animals based on behavioral differences during the forced swim test. She generated two very closely related Wistar substrains characterized by More or Less Immobility during the test (the WMI and WLI lines). Both lines are now fully inbred (>35 F generations). Behaviors of WMI resemble facets of human MDD and anxiety, including depressed mood, disturbed sleep, appetite, etc. Here we describe the full genome sequencing of these two substrains and compare their genomic differences with the goal of nominating strong candidate genes responsible for their marked behavioral differences. DNA was extracted from liver tissues and sequencing was carried out on two platforms: Illumina HiSeq X Ten and Ion Torrent. Reads were mapped to the rat reference genome (rn6). On average, we obtained 27–41 X coverage of the WLI and WMI genomes on each platform. SNPs were identified by using the GATK haplotype caller. The two substrains differ at a total of ~4,400 SNPs. Most are located in noncoding regions but there are a small number of intriguing mutations located in exons (e.g. Pclo) or splicing sites (e.g., Rab1a, Slc01a2, Ryr3, Lyg1, and Nap1l1). We are currently validating and extending these data using linked-read libraries from high molecular weight DNA (100–150 kb, 10X Chromium libraries). This new method will enable us to detect longer range structural variants. We randomly selected 20 SNPs and 10 indels for validation using Sanger resequencing and confirmed 19 SNPs and 9 indels. The minimal genomic differences between these strains provides a unique opportunity to identify causal genes and potentially new mechanisms that modulate depression and related behavioral traits.Boris Tabakoff1, Laura Saba1, Melinda Dwinell2, Mary Shimoyama2, Michal Pravenec3, Robert Williams4
1Skaggs School of Pharmacy & Pharmaceutical Sciences, University of Colorado, Aurora, CO
2The Medical College of Wisconsin, Milwaukee, WI
3Institute of Physiology, Czech Academy of Sciences, Prague, Czech Republic
4The University of Tennessee, Memphis, TN
Status and Composition of the Hybrid Rat Diversity Panel (HRDP)
Systems biology requires appropriate analytical tools, and properly structured model systems to access the power of this approach. Animal models that are useful in applying a systems biologic analysis, should be powerful enough to provide good genomic mapping resolution and be genetically stable for long periods. In this way, they can accommodate the accumulation of data, including the transcriptome, proteome, metabolome, physiology and behavior. A profitable animal model for systems biology is a combination of recombinant inbred strains and classic inbred strains that can capture both mapping power and generalizability on a species perspective. We are establishing a Hybrid Rat Diversity Panel (HRDP) consisting of 96 strains of animals chosen on the basis of genetic diversity. The full HRDP, when completed, will include animals from two recombinant inbred strains (HXB/BXH) and (FXLE/FEXF) and 32 other inbred strains. All of these animals have been genotyped and DNA sequence data is available on many of the strains. The Medical College of Wisconsin will rederived all strains (given funding) and will make animals and tissues available to investigators. At the University of Colorado, we have been gathering transcriptome data on four organs (brain, liver, heart and brown adipose tissue) of the HRDP (RNASeq and exon arrays) and have completed collection of data on half of the strains of animals. Identification and quantitation includes both protein coding and non-coding RNA transcripts. The transcriptome information, including isoforms, has been utilized for network analysis (WGCNA) and single transcript analysis. 3’ UTR analysis allows for mapping of miRNA binding sites. We have instituted a pipeline for systems analysis going from DNA to RNA to physiology/behavior and, even with the current data on half of the HRDP, have realized instructive results. All of our data and analysis tools are available through http://PhenoGen.ucdenver.edu and the Rat Genome Database (RGD). Support by NIAAA, NHLBI and NIDA.Jennifer R Smith, Stanley J Laulederkind, G Thomas Hayman, Shur-Jen Wang, Matthew J Hoffman, Elizabeth R Bolton, Yiqing Zhao, Omid Ghiasvand, Jyothi Thota, Monika Tutaj, Marek A Tutaj, Jeffrey L De Pons, Melinda R Dwinell, Mary E Shimoyama
Rat Genome Database, Department of Biomedical Engineering, Medical College of Wisconsin and Marquette University, Milwaukee, WI, 53226, USA
RGD: data and tools for precision models of human disease
A major challenge for preclinical research is finding, or establishing, a good model for the human disease of interest—one that, more or less, faithfully recapitulates the phenotypic and genetic profile of that disease in the human system. In many cases, canonical model organisms such as rat or mouse are acceptable models, but this is not always the case. As such, the Rat Genome Database (RGD, http://rgd.mcw.edu) has undertaken to incorporate additional mammalian species to allow researchers to leverage a rich dataset across multiple species to find the best model for their needs. In addition to rat, RGD has always offered data for human and mouse for the purpose of cross-species comparisons. Now these have been enhanced with data for long-tailed chinchilla (Chinchilla lanigera), 13-lined ground squirrel (Ictidomys tridecemlineatus), bonobo (Pan paniscus, also known as pygmy chimpanzee), and dog (Canis lupus familiaris). In each case, these species are used as models for human disease, including diseases of the inner and middle ear, retinal diseases, cancer, heart disease, arthritis, autoimmune dysfunction and hypoxia-reperfusion injury. Utilizing the existing robust and adaptable infrastructure, RGD has imported gene records, genomic data and ortholog assignments for these species from NCBI, as well as protein information and Gene Ontology (GO) annotations where available from UniProtKB. Further functional information has been added to these records via the assignment of GO, disease and pathway annotations based on sequence similarity to human, rat and mouse genes. In addition to incorporating this data into the database, work is well underway to expand RGD's suite of analysis tools to include genes from all of these species wherever possible. Chinchilla, dog, bonobo and squirrel JBrowse genome browsers have already been made available at RGD. All four species have also been incorporated into the OLGA advanced search and Gene Annotator tools. Also, although the available data is limited, work is underway to add these species to the InterViewer protein-protein interaction visualizer. This expanded offering of data for multiple species and the analysis tools to easily and efficiently leverage this data gives researchers an excellent resource for discovering precision models for their diseases of interest.Clarissa C. Parker1, Dan Gatti2, Troy Wilcox2, Eric Busch3, Steven Kasparek1, Drew Kreuzman1, Benjamin Mansky1, Sophie Masneuf3, Erica Sagalyn3, Kayvon Sharif1, Dominik Taterra1, Walter Taylor1, Mary Thomas1, Andrew Holmes3, Elissa J. Chesler2
1Department of Psychology and Program in Neuroscience, Middlebury College, VT 05753
2Center for Genome Dynamics, The Jackson Laboratory, 600 Main Street, Bar Harbor, ME 04609
3Laboratory of Behavioral and Genomic Neuroscience, NIAAA, NIH, Rockville MD 20852
Genome-wide mapping of ethanol sensitivity in the Diversity Outbred mouse population
A strong predictor for the development of alcohol use disorders (AUDs) is altered sensitivity to the intoxicating effects of alcohol. Individual differences in the initial sensitivity to alcohol are controlled at least in part by genetic factors. Mice offer a powerful tool for elucidating the genetic basis of behavioral and physiological traits relevant to AUDs; but conventional experimental crosses have only been able to identify large chromosomal regions rather than specific genes. Genetically diverse, highly recombinant mouse populations allow for the opportunity to observe a wider range of phenotypic variation, offer greater mapping precision, and thus increase the potential for efficient gene identification. We have taken advantage of the newly developed Diversity Outbred (DO) mouse population to identify and map narrow quantitative trait loci (QTL) associated with ethanol sensitivity. We phenotyped 778 JAX Diversity Outbred mice (DO) for three measures of ethanol sensitivity: ataxia, hypothermia, and loss of the righting response (LORR). We genotyped a subset of these mice at ~150k markers across the genome and performed high precision QTL mapping using the R program DOQTL. A paired samples t-test indicated that on average, there was a significant and robust decrease in pre-ethanol performance as compared to post-ethanol performance on the Rotarod latency to fall, t(786) = 26.6, p < 0.0001, d = .95. A repeated-measures ANOVA indicated that following ethanol administration, subjects showed significant changes in body temperature over time, F(3.02, 2352.90) = 1098.30, p < 0.0001, η_p^2 = 0.59. During LORR testing, the majority of subjects (87.7%) both lost and regained the righting reflex during the testing period, with duration of LORR ranging from 0 minutes to the cut-off time of 180 minutes (M = 75.9, SD = 52.9). Importantly, we observed tremendous variation in all three traits which enables genetic mapping of naturally occurring genetic variation that is associated with trait variation. We identified four significant QTLs associated with ethanol sensitivity on chromosomes 1, 9, 10, & 16 (-log10pvalue > 6.1). The high genetic precision and phenotypic diversity in the DO may facilitate discovery of previously undetectable mechanisms underlying predisposition to develop AUDs. With the inclusion of RNA-Seq and other molecular profiling we will be able to apply a systems genetic strategy to construct the network of correlations that exist between DNA sequence, gene expression values and ethanol-related phenotypes. This information can in turn be used to identify alleles that contribute to AUDs in humans, elucidate causative biological mechanisms, or assist in the development of putative treatment strategies.David Siefker, Kent Willis, Robert Williams, Lu Lu, Dahui You, and Stephania Cormier
Univ. of Tennessee Hlth. Sci. Ctr.
The influence of genetic diversity on neonatal respiratory syncytial virus Disease
Respiratory Syncytial Virus (RSV) is the number one cause of lower respiratory infection in children and is linked to asthma development later in ltte. After acute infection, some infants develop severe lower respiratory disease that requires hospitalization and can even cause death. This collaborative project aims to identify genetic determinants associated with susceptibility to RSV infection during the neonatal period. Our laboratory has developed a neonatal mouse model of RSV infection to study immunopathology in an age-appropriate manner. Recombinant inbred strains of mice have been extensively sequenced for genetic polymorphisms and are used as a forward genetics approach to identify groups of genes involved in phenotypes of interest, such as disease susceptibility. Here, we utilize the BXD family of recombinant inbred strains of mice with our neonatal RSV infection model to discover genetic loci involved in the susceptibility of neonates to RSV. We show a difference in viral burden in the lungs of neonatal mice between different BXD strains, suggesting a role for genetic variation in RSV susceptibility. Therefore, we have a novel system to identify genetic loci involved in the development of RSV disease. These results will be critical in the development of specific antivirals and vaccines against RSV, which remain elusive despite five decades of research.Molly E. Harmon1, Asmaa A. Sallam1, Slawo Lomnicki2, Stephania A. Cormier1
1Department of Pediatrics, University of Tennessee Health Sciences Center, Memphis, TN
2Department of Environmental Sciences, Louisiana State University, Baton Rouge, LA
Particulate matter exposure effects on aryl hydrocarbon receptor activation and genetic susceptibility of respiratory disease
Exposure to particulate matter (PM) pollution has long been associated with increases or exacerbations of respiratory diseases and is a serious public health concern. PM is a complex mixture of components derived from many sources, including windblown dust and emissions from remediation of Superfund wastes. We are beginning to understand that PM containing environmentally persistent free radicals (EPFRs) may be particularly hazardous to respiratory health. Inhalation exposure to EPFRs is associated with increased airways disease including asthma, but the mechanisms are poorly understood. Our laboratory has shown a role for Th17 cells in EPFR-induced asthma, and because the aryl hydrocarbon receptor (AhR) plays a role in xenobiotic-metabolism as well as in Th17 differentiation, we examined the role of AhR activation with EPFR exposure. In humans, tremendous variability in susceptibility to adverse respiratory effects of PM have been observed. To identify a genetic contribution to adverse respiratory effects of EPFR exposure, we examined expression of the cytochrome p450 enzyme Cyp1a1 (an AhR responsive gene) in recombinant inbred strains of mice known as the BXD family. BXD mice were exposed to EPFRs via oropharyngeal aspiration, lungs were collected at 4hr post exposure, and activation of AhR via expression of Cyp1a1 was analyzed by RT-qPCR. We are in the process of identifying genetic loci and candidate genes that may play a role in determining susceptibility to adverse respiratory effects of EPFR exposure. It is anticipated that such information will identify the most effective preventative targets for individuals who are genetically susceptible and help inform policy decisions.Christian Fischer, Zachary Sloan, Derk Arends and Pjotr Prins
Genetics Browser: a new multi-dimensional web-based exploration tool
In genetics we are dealing with multi-dimensional data. GeneNetwork does QTL mapping, GWAS and even PheWAS. In addition it can correlate QTL, compute simple phenotype correlations, as well as more advanced correlation trait loci (CTL), weighted correlation network analysis (WGCNA) and bayesian network analysis. For the researcher, unfortunately, this means mining lists of correlated data and only having a 'legacy' two dimensional genome browser or QTL map viewer to explore the genome.
For GeneNetwork we are developing a new multi-dimensional web-based exploration tool. Starting from Biodalliance (BD), an interactive, web-based two-dimensional genome browser, we are adding multi-dimensional exploration functionality with Cytoscape.
Areas of interest in the network data can be found by exploring the genomic data. Hotspots in the genome can be further explored in the Cytoscape graph, going back and forth between genome, phenotypes, experiments and even external data, such as genome annotation and relevant publications.
CF is funded by Google Summer of Code Projects (2016 and 2017) under the auspices of the Open Bioinformatics Foundation (OBF).
Suheeta Roy1, Pooja Jha7, Evan G. Williams8, Maroun Bou Sleiman7, Hyeonju Kim2, Megan K. Mulligan1, Khyobeni Mozhui2, Jesse Ingels1, Casey Bohl1, Melinda McCarty1, Jinsong Huang1, Hao Li7, Richard A. Miller3, James F. Nelson4, J. Randy Strong5, David E. Harrison6, Saunak Sen2, Lu Lu1, Johan Auwerx7, Robert W. Williams1
1Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Center, Memphis, TN 38163, United States
2Department of Preventive Medicine, University of Tennessee Health Science Center, Memphis, TN 38163, United States
3Department of Pathology and Geriatrics Center, University of Michigan, Ann Arbor, MI, 48109-2200, USA
4Department of Physiology and Barshop Center for Longevity and Aging Studies, The University of Texas Health Science Center at San Antonio, San Antonio, TX, 78229, USA.
5Geriatric Research, Education and Clinical Center and Research Service, South Texas Veterans Health Care System, Department of Pharmacology, The University of Texas Health Science Center at San Antonio, San Antonio, TX, 78229, USA.
6The Jackson Laboratory, Bar Harbor, ME, 04609, USA
7Laboratory of Integrative and Systems Physiology, École Polytechnique Fédérale de Lausanne, Lausanne CH-1015, Switzerland
8Department of Biology, Institute of Molecular Systems Biology, ETH Zurich, Zurich CH-8093, Switzerland
Genetic analysis of longevity in diverse cohorts of mice: Influence of Diet and Drugs
Background:Diet, drugs, and environmental factors modulate lifespan. We are using complementary molecular and genetic methods to evaluate the impact and interactions of genetic variants, diet, and drugs on aging, weight gain, and metabolism. BXD mice maintained on two diets and a 4-way F2 intercross (the NIA Interventional Testing Program- www.nia.nih.gov/research/dab/interventions-testing-program-itp) are used as cohorts. Both cohorts segregate for ~5–7 million variants, making them ideal for the analysis of gene-by-diet and gene-by-drug interactions that modulate metabolism and lifespan.
Methods:(1) We measured longevity as a function of diet and weight gain in females from both parental strains C57BL/6J and DBA/2J, and ~70 BXD strains on a standard chow diet (CD, 6% calories from fat) or a high fat diet (HFD, 60% calories from fat). A subset of animals were sacrificed at ~7, 12, 18, and 24 months for ongoing multi-omics analyses (abstract by Williams EG et al.) All of these strains are being sequenced at ~30X using 10X linked read libraries. (2) The NIA ITP program has measured longevity and weight gain in ~14,000 UM-Het3 F2 intercross mice [(C57BL/6JxBALB/cByJ)x(C3H/HeJxDBA/2J)] raised in carefully balanced subsets in Michigan, Texas, and Maine. Subsets of ITP cases were treated with compounds suspected to have effects on lifespan—from aspirin to rapamycin. Here we report data only for control cases.
Results:(1) By 500 days BXDs on HFD gained 5X more weight than those on CD (5.2 vs. 27.6 g). Lifespan was shortened by ~90 days (589 ± 8 days, n = 540; CD 672 ± 9, n = 508), equivalent to 6-7.5 years in humans. Longevity under the two diets correlates poorly (r = 0.37). Remarkably, baseline weight and weight gain within diet do not correlate with longevity; demonstrating that the diet itself, rather than weight gain modulates longevity in female BXDs. Preliminary QTL mapping of CD and HFD BXD cohorts yielded suggestive QTLs for lifespan (GeneNetwork Traits 18441 and 18435). Matched omics analysis of liver and other tissues is in progress to help us dissect molecular networks modulating metabolic aging. (2) Preliminary analyses based on a subset of 2400 control mice from the ITP study highlight a suggestive longevity QTL on Chr 15 (LOD score 4.66) in male mice. QTL scans reveal that the genetic determinants of longevity and weight gain are sex-specific. Additional samples will provide a powerful panel to study the genetics of longevity in relation to sex and metabolic status.
Hemant Gujar1, Nicholas C. Wong2, Jane W. Liang1, Khyobeni Mozhui1,3
1Department of Preventive Medicine, University of Tennessee Health Science Centre, Memphis, Tennessee, 38163, USA
2Department of Paediatrics, The University of Melbourne, Royal Parade, Parkville, Victoria 3052, Australia.
3Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Centre, Memphis, Tennessee, 38163, USA
Conserved probes in the human MethylationEPIC microarray to profile DNA methylation in mice
The Illumina Infinium MethylationEPIC BeadChip provides a robust and efficient platform to profile DNA methylation at over 850000 CpG sites in the human genome. Model organisms such as mice do not benefit from a similar cost-effective and convenient microarray. Here we examine the utility of the human array as a means to measure DNA methylation in mouse samples. We aligned the 50-mer probe sequences to the mouse genome to identify probes that target conserved CpG sequences. We then examined the performance of these conserved probes in mouse samples, and compared it with relative methylation detected by affinity-base DNA sequencing of methyl-CpGs (MBD-seq). Mouse samples consisted of 11 liver DNA from two strains, DBA/2J and C57BL/6J, that varied widely in age. Linear regression analysis was applied to detect differential methylation as a function of mouse strain and age. In total, 13665 probes (1.6% of total) align with high confidence to the mouse genome, and are highly enriched in exonic regions, and CpG islands that are upstream of genes This set provides a reliable measure of DNA methylation in mice with similar beta-value (β) distribution as in human samples. At a Benjamini Hochberg threshold of p ≤ 0.05, 81 probes detected differential methylation between strains and two probes detected change in methylation with age. The comparison between the microarray and sequencing data shows that methylation levels detected by the two technologies are highly concordant (Pearson correlation R = 0.70, p < 0.0001). When grouped by methylation β scores, the linear correlation is stronger for hemi-methylated CpGs (i.e., 0.3 ≤ β ≤ 0.7, R = 0.46), followed by hypomethylated CpGs (β < 0.3, R = 0.18), and hypermethylated CpGs (β > 0.7, R = 0.04). The poor correlation of hypermethylated CpGs is likely due to low quantitative sensitivity of the MBD-seq at hypermethylated sites. Overall, the MethylationEPIC probes perform better at detecting differential methylation. In conclusion, we find that only a small subset of the 850K probes target conserved sites. However, these provide reliable assay of DNA methylation, and may have higher quantitative sensitivity than the alternative affinity-based sequencing.Jinsong Huang1, Lu Lu1, Zhousheng Xiao2, Bing Zhang3, Jing Wang3, Danny Arends4, Weikuan Gu5, Leigh Darryl Quarles2, Robert W. Williams1
1Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Center, Memphis, TN, USA
2Department of Medicine, University of Tennessee Health Science Center, Memphis, TN, USA
3Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA
4Humboldt University, Berlin, Germany
5Department of Orthopaedic Surgery and Biomedical Engineering, University of Tennessee Health Science Center, Memphis, TN, USA
Genetic modulation of bone microtraits: Joint QTL, gene expression and gene ontology (GO) analysis
BackgroundBone quality and quantity are polygenic and complex traits modulated by a large number of genetic and environmental factors and characterized by many adaptive and age-related changes. There is now greater appreciation of the power of experimental rodent models to study complex traits and to evaluate candidate genes controlling different aspects of bone structure and function. In this study, we have measured complex bone microtraits in a cohort of BXD recombinant inbred (RI) strains, computed heritabilities, mapped sets of quantitative trait loci (QTLs) that influence complex bone traits, and devised a new method to rank candidate genes.
Materials and MethodsWe used high-resolution microCT (Scanco μCT40) to measure a set of 50 bone phenotypes from femur and tibia of 61 BXD strains and both parental strains, C57BL/6J and DBA/2J. We entered age-normalized data into GeneNetwork (GN18130-18279) for mapping and to define positional candidate genes. We also performed gene ontology (GO) analysis using a hypergeometric test [Wang et al, PMID: 23703215] and a large bone gene expression dataset [Farber et al, PMID: 23300464] to compute GO-associated “boniness scores” for all positional candidates, and established a scoring system to generate a summary candidate score for all genes. This GO scoring method was used in combination with other conventional criteria (e.g. SNP scores of coding DNA variants and a cis-regulation score) to more objectively assess associations of candidate genes with bone biology. We have defined a bone “ignorome”— the set of uncharacterized genes likely to be important in skeletal system biology and function [Pandey et al, PMID: 24523945].
Results and DiscussionThe majority of bone microtraits have heritabilities that range from 40% to 64% in both sexes. We mapped 17 QTLs, 11 for femur and 6 for tibia, based on 25 male, 25 female, and 25 sex-averaged bone traits. We defined a total of 1698 positional candidates within 1.5 LOD confidence intervals. Using a scoring system with scale from 1 to 10, we systematically ranked candidate genes that may contribute to variation in bone size and architecture, and highlighted a subset of 219 candidates with scores greater than 4. Approximately 20% of these genes are known to be related to bone biology, including bone matrix homeostasis, osteoblasts/osteoclasts regulations, and some types of genetic bone disorders. However, the remaining 80% genes have no known association in the literature with bone biology. A few of these strong bone ignorome candidates with scores of 5 or higher include Obsl1, Ifi205, Zbtb6, Mgmt, Cfap46, and Greb1. This innovative GO-based analysis method of complex traits provides an additional computational approach to uncover gene functions in the skeletal system and in other tissues and organs.
Diana X. Zhou1, Jessica A. Baker2, Kristin M. Hamre2, Junming Yue3, Byron C. Jones1, Robert W Williams1, Melloni N. Cook4, Lu Lu1
1Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Center, Memphis, TN 38163, United States
2Department of Anatomy and Neurobiology, University of Tennessee Health Science Center, Memphis, TN 38163, United States
3Department of pathology, University of Tennessee Health Science Center, Memphis, TN 38163, United States
4Department of Psychology, University of Memphis, Memphis, TN 38152, United States
The Effect of Alcohol on the Differential Expression of Cluster of Differentiation 14 Gene, Associated Pathways, and Genetic Network
Alcohol consumption affects human health in part by compromising the immune system. Cd14 (cluster of differentiation 14) is a gene vital in the immune system’s inflammatory cascade. In this study, we combine the power of BXD RI strains of mice with systems genetics approaches to determine the genetic regulation of Cd14, identify a genetic network for Cd14, and explore genes correlated to Cd14 and pathways through which Cd14 is involved in ethanol response. Hippocampal gene expression data were generated from BXD mice treated with saline or ethanol (12.5% v/v, 1.8 g/kg i.p.). The Cd14 gene expression showed significant changes among the BXD strains after ethanol treatment, and eQTL mapping revealed that Cd14 is a cis-regulated gene. We also identified eighteen ethanol-related phenotypes correlated with Cd14 expression related to either ethanol responses or ethanol consumption. Pathway analysis was performed to identify possible biological pathways involved in the response to ethanol and Cd14. We also constructed a genetic network for Cd14 using the top 20 correlated genes and present several genes possibly involved in Cd14 and ethanol responses based on differential gene expression. In conclusion, we found Cd14, along with several other genes and pathways, to be involved in ethanol responses in the hippocampus, such as increased susceptibility to lipopolysaccharides and neuroinflammation.Prasun Anand1 and Pjotr Prins2
1Birla Institute of Technology and Science, Goa, India 2University Medical Center Utrecht, The Netherlands
Faster Linear Mixed Models (LMM) for online GWAS omics analysis
We are writing, refactoring, and optimizing linear mixed model (LMM) code in the D programming language for near- realtime genome scans on GPUs and clusters. The new package is called Faster-LMM-D (github.com/prasunanand/faster_lmm_d). Code handles complex admixture, large numbers of cofactors, and should be able to map using large GWAS-type populations. Our goal is that Faster-LMM-D be sufficiently fast for on-the-fly mapping and modeling of large omics data sets using powerful server-side hardware. This work follows up on FaST-LMM by Lippert and colleagues (PMID: 21892150), and a Python implementation (pylmm) by Nick Furlotte and colleagues (github.com/nickFurlotte/pylmm). Pylmm is already functionally part of GeneNetwork 2 (http://gn2.genenetwork.org/). By writing the new version in D we have already achieved significant speedups and we are currently adding methods to efficiently use high-core count CPU and GPU hardware, including NVIDIA GPUs and Intel Xeon Phi processors. Test implementations of Faster-LMM-D are running on GeneNetwork 2 staging servers and will be available for general use in 2017-2018.Acknowledgements: We thank NIGMS R01GM123489 and the UT CITG for support of GeneNetwork.
Alharbi NHJ, McBride MW, Padmanabhan S, Graham D
Institute of Cardiovascular and Medical Sciences, College of Medical, Veterinary and Life Sciences, University of Glasgow, 126 University Place, Glasgow G12 8TA, United Kingdom
Investigating a novel pathway underlying hexadecanedioate induced blood pressure elevation
Background: A putative novel pathway for blood pressure regulation involving the dicarboxylic acid hexadecanedioate was identified in a study associating blood pressure and mortality outcomes with fasting blood metabolites [1]. The functional role of hexadecanedioate on blood pressure elevation and vascular reactivity was confirmed by oral administration of hexadecanedioic acid to Wistar Kyoto (WKY) rats [1]. The present study aimed to characterize the metabolic effects of hexadecanedioic acid administration in WKY rats, and assess hemodynamic changes after modulation of endogenous hexadecanedioate levels by perturbing the ω-oxidation pathway in the stroke-prone spontaneously hypertensive rat (SHRSP).
Methods: Eleven-week-old male WKY were treated orally with hexadecanedioic acid (250 mg/kg per day; n=5) or vehicle (n=5) for 3 weeks. At sacrifice tissues (aorta, heart, brain, adipose, kidney, and liver) were harvested and global metabolic profiles analysed by UPLC-MS/MS (Metabolon). Disulfram, an inhibitor of the ω-oxidation pathway enzyme, aldehyde dehydrogenase (ALDH), was administered to twelve-week-old male SHRSP rats by intraperitoneal injection (25 mg/kg/day, n=6), or vehicle (n=6) for 14 days. Blood pressure was assessed by tail plethysmography and mesenteric arteries were used to assess vascular function by wire myography.
Results: Treatment with hexadecanedioic acid increased hexadecanedioate levels in all tissues tested except the brain in WKY rats. Metabolomic analysis identified increased fatty acid metabolites (e.g. acylcarnitines) in heart, long-chain fatty acids and ketone body (β-hydroxybutyric acid) in kidney, and dicarboxylate fatty acids (tetradecanedioate and octadecanedioate) in adipose tissue from hexadecandioic acid treated rats indicating an impairment of fatty acid beta-oxidation and a shift towards peroxisomal ω-oxidation. SHRSP rats showed a significant reduction in blood pressure after treatment with the aldehyde dehydrogenase antagonist, disulfiram, for two weeks (∆SBP, 23.0±4.5 mmHg; P=7.47x10-5). Mesenteric resistance arteries from disulfram treated SHRSP demonstrated a shift to the right in the contractile response curve to noradrenaline, indicating reduced vascular sensitivity compared to control vessels.
Conclusion: Exogenous administration of hexadecanedioic acid in addition to increasing blood pressure impacts several metabolic readouts including changes related to fatty acid metabolism. Importantly, we demonstrate that inhibition of the ω-oxidation pathway enzyme, aldehyde dehydrogenase, results in a significant lowering of blood pressure in the hypertensive SHRSP rat. The ω-oxidation pathway is a target for further research to elucidate the mechanism by which hexadecanedioate affects blood pressure.
Robert W. Williams, Danny Arends, Megan K. Mulligan, Evan G. Williams, Cathleen Lutz, Alicia Valenzuela, Casey J. Bohl, Jesse Ingels, Melinda S. McCarty, Arthur Centeno, David G. Ashbrook, Johan Auwerx, Lu Lu
The expanded BXD family: A cohort for experimental systems genetics and precision medicine
We have expanded the size of the BXD family of recombinant inbred strains two-fold—from 70 to ~150 strains. Roughly 100 strains are now available from the Jackson Laboratory and a total of 116 are beyond F20, and ~24 are between F10 and F20. We have generated updated genotypes for new strains (n = 198, including ~50 that are extinct) using the GigaMUGA array. The BXD family segregates for ~5 M common variants (PMID:268330985) and a large number of still undefined rare variants unique to each line and subfamily. Genetic maps based on ~7500 informative SNPs conform to consensus physical and genetic maps and have an adjusted length corresponding to 1400 cM in a standard cross. Recombinations between parental haplotypes have been located with a precision of ~200 kb. The family is now being sequenced at ~35x (Illumina HiSeq X10) using high molecular weight linked-read libraries (10X Chromium). We expect to have near final genometypes for 150 extant strains in 2018, along with a compendium of rare mutations. Both the sequence data, de novo assembly, and infinite marker maps will enable more confident modeling and testing of genome<–>phenome relations and complex experimental models of GXE interactions. Based on empirical data and analysis of cis eQTLs, it is already practical to achieve mapping precision of under ±2.0 Mb over much of the genome using 100 BXDs. We estimate that the asymptotic precision of the family is about ±500 kb for Mendelian loci with LOD>10 (Pandey, 2014, PMID:25172476). With a sample size of >100 genomes, the BXD family is also sufficiently large to detect some types of two-locus epistatic interactions.
The BXD family has a remarkably deep and well-structured phenome available as www.genenetwork.org that includes more than 70 gene expression, proteome, metabolome and metagenome data sets and ~5000 quantitative and experimental "classic" phenotypes. As a result this family is now an unrivalled resource for testing networks of causal and mechanistic relations among clinical phenotypes and millions of molecular and organismal traits, including metabolic syndrome, infection, addiction, and neurodegeneration, and longevity. Precisely matched cohorts can be raised in under different conditions to study gene-by-environmental interactions, epigenetic modifications, replicability, and robustness of genome-to-phenome relations. The BXDs are already well suited for "reverse" phenome-wide association studies of loci and gene variants. In short, the BXDs are now a mature resource of experimental predictive biology and even health care.
Acknowledgements: The production of the BXDs is supported by the UTHSC Center for Integrative and Translational Genomics to RWW and colleagues. The importation, rederivation, and distribution of BXD strains at The Jackson Laboratory is supported by NIH P40 ODS011102 to CL and colleagues.
Spencer Mahaffey, Laura Saba, Lauren Vanderlinden, Yinni Yu, Paula L. Hoffman, Boris Tabakoff
University of Colorado School of Pharmacy and Pharmaceutical Sciences and School of Medicine, Aurora, CO 80045, USA
The rat based pipeline for systems genetic analysis
One of the most important resources for systems genetic studies is a genetically stable (isogenic) panel of strains, which allows for fine mapping of complex phenotypes (QTLs) and provides statistical power to identify loci which contribute nominally to the phenotype. Often, rats are preferred over mice for physiologic and behavioral studies because of their larger size and more distinguishable anatomy (particularly for their CNS). The HRDP is such a panel of rat strains which combines two recombinant inbred panels (the HXB/BXH, 30 strains; the LEXF/FXLE, 33 strains and 33 more strains of inbred rats which were selected for genetic diversity, based on their fully sequenced genomes and/or thorough genotyping). The HRDP will provide a genetically stable group of animals whose genomes are well-sequenced (30x) and the total RNA (coding, non-coding, large, small) of brain, liver and heart will be characterized by RNAseq. The RNA will be mapped to the genome, annotated as possible, and assigned to modules and networks by use of WGCNA. The QTLs for individual transcripts and module QTLs will also be available for integration with phenotypic QTLs. The HRDP will be particularly suited to investigations of predisposition or susceptibility loci for physiologic and pathologic phenotypes. Although the completion of the HRDP is a work in progress, sufficient work has been performed and analyzed to provide confidence that this resource will well serve the addiction research community. The genomic sequence of the progenitors of the HXB/BXH panel has been completed and is available, as is the imputed sequence of the 30 RI strains of this panel. The genomic sequence of an additional 10 inbred strains of HRDP is also available. RNAseq data for brain, left ventricle of heart, and liver are also available as are data on cell-specific RNA expression in liver (hepatocyte, Kupffer, stellate and endothelial cells). All data have been quality controlled, normalized and cleaned and are available in this form, or as raw data. The data are also available in an analyzed format, including visualization formats using genome tracks and network/QTL visualization tools. The portal to access all information and tools is http://phenogen.ucdenver.edu. A number of publications have been generated using the data on just the HXB/BXH RI strains and this work has identified a viable pipeline for incorporating genomic, transcriptomic, and phenotypic data into a systems analysis of biologic diversity leading to individual differences in predisposition to behavioral and physiologic phenotypes (supported by R24 AA013162 and the Banbury Fund).Andrew B. Stiemke, Monica M. Jablonski
Systems Genetics of Optic Nerve Axon Death
Purpose: To identify strong gene candidates for the modulation of axon death in retinal ganglion cells.
Methods: 73 different BXD strains, parents, and F1 crosses were used for ocular nerve harvesting. All animal care followed all required guidelines. The optic nerves were sectioned and stained with 1% p-phenylenediamine for counting using ImagePad software. The obtained data were uploaded to GeneNetwork (www.genenetwork.com) for systems genetics analyses. After observing a peak quantitative trait locus, a set of criteria, including Pearson correlation analysis, gene expression, and known functioning in a network that could modulate axon death, were followed to first obtain and then narrow down potential gene candidates.
Results: Of 214 initial gene candidates, four genes, Cdc42bpb (CDC42 Binding Protein Kinase Beta), Serpina3g (Serine protease inhibitor A3G), Tmem179 (Transmembrane Protein 179), and Apopt (Apoptogenic 1, Mitochondrial), passed all applied criteria.
Discussion: The final four potential gene candidates can be sorted into two main categories, axonal influence and ganglion cell influence. Cdc42bpb, Serpina3g, and Apopt have direct axonal influence, while Tmem179 participates in an intracellular cGMP activated cation channel activity network, which could modulate retinal ganglion cell activity. Further testing, such as immunohistochemistry or gene knockdown, is needed to further confirm or rule out the remaining gene candidates.
Hanifa J. Abu-Toamih Atamni1, Maya Botzman2, Richard Mott3, Irit Gat-Viks2, Fuad A. Iraqi1
1Sackler Faculty of Medicine, Tel-Aviv University, Tel-Aviv, Israel
2Faculty of Life Sciences Tel-Aviv University, Tel-Aviv, Israel
3Department of Genetics, University College of London, London, UK
Mapping novel genetic loci associated with female liver weight and fattiness variations in Collaborative Cross mice
Introduction:Liver features are complex traits controlled by polygenic factors that differs between and within populations. Dissecting genetic architecture underlying these variations will facilitate the study of multiple hepatic disorders.
Aims:Mapping Quantitative Trait Loci (QTL) underlying liver weight and fattiness phenotypic variations in Collaborative Cross (CC) mice.
Materials and Methods:506 mice of 39 different CC lines (both sexes) at age of 20 weeks old assessed for liver weight (electronic balance) and fattiness (DEXA scanner). Genomic DNA of the CC lines was genotyped.
Results and Conclusions:Liver weight and fattiness varied significantly (P <0.05) between CC lines and sexes. QTL mapping was significant only for females. Liver weight QTL mapped on Chr8 (88.61-93.38 Mb) and three suggestive QTL mapped at Chr4, Chr12 and Chr13, named LWL1- LWL4 (Liver Weight Locus1-4). Liver fattiness QTL mapped on Chr17 (3 Mb), Chr18 (2 Mb), and less significant on Chr4 (2 Mb), named Flal1-Flal3 (Fatty Liver Accumulation Locus 1-3), respectively. Our findings demonstrates , for the first time, utilization of the CC lines for mapping QTL associated with baseline liver phenotypic variations. Concluding that baseline liver weight and fattiness differ between sexes, whereas mapping these genetic components will contribute to the study of hepatic abnormalities (growth retardation/ hepatomegaly/ Non-Alcoholic Fatty Liver disease).
Hanifa J. Abu-Toamih Atamni1, Georgia Kontogianni2, Richard Mott3, Heinz Himmelbauer44, Hans Lehrach5, Aristotele Chatziioannou2, Fuad A. Iraqi1
1Department of Clinical Microbiology and Immunology, Sackler Faculty of Medicine, Tel-Aviv University, Tel-Aviv, Israel
2Institute of Biology, Medicinal Chemistry & Biotechnology, Athens, Greece
3Department of Genetics, University Collage of London, UK
4Centre for Genomic Regulation (CRG), Barcelona, Spain
5Department of Vertebrate Genomics, Max Planck Institute for Molecular Genetics, Berlin, Germany.
Hepatic gene expression in health and disease using the collaborative cross mouse genetic reference population
Introduction: Hepatic gene expression known to differ between health and disease in T2D and Metabolic syndrome (MetS). Study in-depth of these variations will contribute to the development of gene-targeted therapies.
Aims: Assess diet-induced hepatic gene expression of susceptible versus resistant CC lines.
Materials and Methods: Next generation RNA-seq for 84 livers of diabetic and non-diabetic mice of 43 CC lines (both sexes) following 12 weeks High-Fat Diet (42 % fat).
Results and Conclusions: Hepatic gene expression differ in diabetic versus non-diabetic with sex effect. Overall population 601 genes, differentially expressed (DE) and 470 alternatively spliced (AS). Females, 718 genes DE and 621 AS, Males 599 genes DE and 479 AS. Top prioritized DE candidate genes were; overall Lepr; Ins2; Mb; Ckm; Mrap2; Ckmt2; Pttg1; Reg1, for females only Hdc; Serpina12; Socs1; Socs2; Mb, for males only Serpine1; Mb; Ren1; Slc4a1; Atp2a1. Sex-differences revealed 193 DE genes and 437 AS in health (Top: Lepr; Cav1; Socs2; Abcg2;Col5a3), while 389 genes DE and 423 AS between diabetic females versus males (Top: Lepr; Clps; Ins2;Cav1;Mrap2). Altogether, emphasizing genetic complexity of T2D and MetS.
Hanifa J. Abu-Toamih Atamni1, Yaron Ziner2, Richard Mott3, Lior Wolf2, Fuad A. Iraqi1
1Department of Clinical Microbiology and Immunology, Tel-Aviv University, Tel-Aviv, Israel.
2The Blavatnik School of Computer Science, Sackler Faculty of Medicine, Tel-Aviv University, Tel-Aviv, Israel.
3Department of Genetics, University Collage of London, London, UK.
Glucose tolerance female-specific QTL mapped in collaborative cross mice
Introduction:Type-2 diabetes (T2D) is a complex metabolic disease characterized by impaired glucose tolerance. Despite environmental high risk factors, host genetic background is a strong component of T2D development.
Aims:Mapping Quantitative Trait Loci (QTL) underlying impaired glucose tolerance phenotypic variations in Collaborative Cross (CC) mice.
Materials & Methods:501 mice of 58 novel highly genetically diverse CC lines maintained on high-fat (42 % fat) diet for 12 weeks followed by 180 min of intraperitoneal glucose tolerance test (IPGTT). Values of Area under curve (AUC) for glucose at zero and 180 min (AUC0-180) were calculated and CC lines genotyped for QTL mapping.
Results and Conclusions:AUC0-180 varied significantly between the CC lines and sexes. Nevertheless, QTL was significant only for females. Female sex-dependent QTL (2.5 Mbp) mapped on Chr8 (32–34.5 Mbp), Gene browse of the genomic interval reveals reported QTL for body weight/size, genes involved in immune system, and two main protein-coding genes involved in the Glucose homeostasis, Mboat4 and Leprotl1. In conclusion, genetic factors controlling glucose tolerance differ between sexes requiring independent studies for females and males toward T2D prevention and therapy.
n Memoriam: John K. Belknap and Gerald E. McClearn, Ph.D.
Founders of Quantitative Behavioral Neurogenetics and Genetics of Drug Use and Abuse
We lost two greats in genetics earlier this year—John Belknap and Gerald McClearn. In different ways that founded and then advanced our understanding of the genetic modulation of a wide range of behavioral, neuro
John Belknap: 1943-2017
We are sad to announce the sudden and unexpected death of John Belknap earlier this year. John was Professor of Behavioral Neuroscience at OHSU and a Senior Research Career Scientist at the VA Portland Health Care System. He earned his BA, MA and PhD (1971) in Psychology from the University of Colorado in Boulder. He was one of the first graduates of both the new Biopsychology program and the new Institute for Behavioral Genetics, where he studied with a pioneer in the field, Dr. Gerald E. McClearn. After a postdoctoral fellowship and a position as Assistant Professor of Psychology at the University of Texas in Austin, John moved to the Pharmacology Department at the University of North Dakota School of Medicine. John was Acting Chair of that department before moving to Portland in 1988.
John epitomized the pure basic scientist and was fascinated with data—the bigger the dataset, the better. He loved nothing better than helping anyone who asked to extract patterns and meaning from complicated data, a skill at which he was adept. His mouse behavioral laboratory work focused on studies of genetic contributions to the effects of drugs of abuse. He had been continuously funded by the NIAAA, NIDA and the VA since 1988 and was the heart of the original collaborative efforts underlying the Portland Alcohol Research Center (and, more recently, the Methamphetamine Abuse Research Center). His quantitative sophistication led many of his wide network of collaborators from Portland and many other research centers gently into the era of behavioral genomics. John was one of the pioneers in developing and proving the idea that behavior was a complex trait, an idea now widely accepted and the forerunner of behavioral GWAS. However despite his many contributions, John never looked for the limelight; he was the rare quiet scientist, despite his fame.
Besides his passion for data, he loved sailing and anything maritime, including the books of Patrick O’Brian. He was an astoundingly knowledgeable amateur astronomer. He loathed anything that even faintly resembled bureaucracy, and avoided it religiously. His gentle presence will be sorely missed by his former students, laboratory assistants, and all those here and elsewhere who knew him. Planning discussions for a suitable event to commemorate his scientific career are underway, and details will follow at a later time.
AF Gileta1, CJ Fitzpatrick2, TE Robinson3, AA Palmer1
1Department of Human Genetics, University of Chicago, Chicago, IL
2Department of Psychiatry, 3Department of Psychology, University of Michigan, Ann Arbor, MI, USA.
Genome-wide association study for the propensity to attribute incentive salience to reward cues in outbred rats.
Incentive salience refers to the motivational value attributed to a reward-predicting stimulus, which causes it to become desired and able to bias attention and elicit approach. In the case of addiction, reward-associated stimuli (conditioned stimuli) that acquire incentive salience can trigger powerful craving and motivate drug-seeking/taking behaviors. Pavlovian Conditioned Approach (PCA) is a behavioral paradigm used to reliably quantitate the propensity of rats to approach and interact with a reward-paired cue. This conditioned response, known as sign-tracking, reflects the extent to which the cue is attributed with incentive salience. This trait has been shown to be both highly variable and heritable in Sprague-Dawley (SD) rats. Over the past 4 years, our collaborators phenotyped a cohort of ~4,000 outbred SD rats from multiple vendors for PCA. We are using this cohort to perform the first large-scale genome-wide association study (GWAS) for the attribution of incentive salience. To obtain genotypes for the rats, we utilize double digest genotype-by-sequencing (ddGBS), a reduced-representation sequencing approach we optimized for this study. A subset of these rats (n=80) are have also been whole-genome sequenced to assay the standing genetic variation and linkage structure in the component subpopulations. Due to the extensive structure observed in SD rat populations, association analyses will be performed using mixed models containing a random effect term with a genetic relatedness matrix estimated from the SNP genotype data. The marker data will also be used to precisely estimate the narrow-sense heritability of this trait.Vivek Kumar, Brian Geuther, Kai Fox, Sean Deats
The Jackson Laboratory, Bar Harbor, ME 04609
Quantitative genetics of serial action coding by the mammalian nervous system
Rule-governed sequences of action are critical for many behaviors. This process of serial action coding is coordinated by the basal ganglia, and its deficits lead to numerous diseases. Here we use mouse grooming as a model to study the genetic basis of serial action coding. We use deep convolutional neural networks for general purpose mouse tracking and recognition of grooming behavior. We are accurately able to detect grooming, and present a 55-strain survey of mouse grooming in the open field. To the best of our knowledge, this represents the largest analysis of rodent grooming behavior. We show that total time grooming and grooming bout length is highly heritable. We carry out genome wide association mapping using PyLMM and detect significantly associated SNP clusters for grooming phenotypes. Initial analysis of genes within the associated intervals indicate several that have been previously linked to abnormal grooming. We are currently modifying our methods to detect syntactic chain grooming. The methodology we have established is highly flexible and applicable to many other behaviors.Laurence Tecott, Giorgio Onnis
University of California San Francisco Department of Psychiatry, San Francisco, CA 94158
A quantitative systems approach reveals fundamental principles of spontaneous behavioral organization exquisitely sensitive to genes and diet
We describe a principled analytical approach that reveals the structure of spontaneous mouse home cage behavior and unprecedented precision in behavioral phenotyping. We have defined, validated and automated detection of "Inactive States", periods in which energy-conserving behaviors are expressed exclusively at a single nest location. These alternate with "Active States" in which behaviors such as exploration, feeding and drinking occur with characteristic diurnal and sequential patterns. Using a large multimodal dataset obtained from 16 genetically diverse inbred strains, we demonstrate these spatiotemporal patterns to be robust generalizable features of Mus musculus behavior. Application of this approach reveals a series of novel insights into the manner in which the CNS organizes and coordinately regulates the diverse behaviors expressed spontaneously in the mouse home cage. Moreover, we find that behavioral patterns are remarkably sensitive to strain. Classical supervised machine learning using Active/Inactive State features provides 99% cross-validated accuracy in genotyping animals using behavioral data alone. Additional insights into behavioral regulation are achieved by defining and quantifying bouts of feeding, drinking and movement that occur within ASs. These have been particularly useful for studying behavioral determinants of energy balance and obesity. We provide evidence that standard food intake and locomotor activity measures are relatively insensitive phenotyping tools. This is likely attributable to redundancies in the neural mechanisms regulating energy balance. By contrast, we find that fine-grained "behavioral dissections" can reveal marked gene- and diet-induced phenotypic alterations in lower-level behavioral elements and the manner in which behavioral compensatory mechanisms are recruited to blunt their disruptive effects on feeding and physical activity levels.
B.C. Jones1, L. Lu1, M.K. Mulligan1, S.A. Cavigelli2, W. Zhao1, R.W. Williams1, E. Terenina3, P. Mormède3
1Univ. of Tennessee Hlth. Sci. Ctr, Memphis, TN
2Penn State Univ. Univ. Park, PA
3INRA UMR1388, Castanet-Tolosan, France
Forward genetic analysis of initial and subsequent consumption of ethanol in a large family of genetically diverse strains of mice
Heritable differences in alcohol use disorders have been recognized for several decades. Many animal models have been employed to define genes variants and biochemical networks that predispose individuals to alcohol-related problems. This includes approach, initial consumption, change or elevation in the amount consumed, and dependence. Few animal models exist that address individual differences in initial exposure, consumption, and change following exposure. To this end, we tested female mice from 36 BXD recombinant inbred strains for initial and continuing alcohol consumption using the drinking in the dark (DID) method. All animals were between 60 and 80 days of age at the start of the study. DID follows a 4-day routine on a weekly schedule. On days 1–3 (Tues–Thurs in our case) water was removed 3h after lights out and replaced for 2h with alcohol (20% v/v from 95% EtOH). On the 4th day, treatment was repeated, but animals had 4h exposure. For this study, we followed this routine for five weeks. For the first 2h exposure, we observed wide variation in EtOH consumed from a low of 1.08 to 3.20 g/kg. For the first 4h exposure we observed a similar wide variation from 2.50 to 6.73 g/kg across 36 strains. The correlation for consumption between these exposures was r = 0.69, p <0.01. Mapping of strain variation in DID for both exposures revealed the same suggestive locus near the telomere of chromosome 1. A possible candidate gene is Gm821, gene model 821 (NCBI). This gene is cis-regulated and its expression correlates with alcohol consumption r >0.60, p <0.01. For the 5th 4h exposure (following 5 weeks of DID), the range of consumption was 3.0 to 7.2 g/kg. The correlation between the first and fifth 4h DID was r = 0.62, p <0.01. For this trait, we detected suggestive loci on chromosomes 16, 18 and 19, but no strong candidate genes. In comparing the 5th 4h exposure to the 1st exposure, ten strains increased their consumption over the 5 weeks, but no strains decreased their consumption. Supported in part by USPHS Grant AA 02951Goldberg LR1, Kirkpatrick SL1, Luong AM1, Luttik KP1, Wu J1, Reed ER1, Jenkins DF1, Kelliher JC1, Crotts S1, Yazdani N1, Johnson WE2, Mulligan MK3, Bryant CD1
1Laboratory of Addiction Genetics, Department of Pharmacology and Experimental Therapeutics and Psychiatry, Boston University School of Medicine
2Department of Medicine, Division of Computational Biomedicine, Boston University School of Medicine
3Department of Genetics, Genomics, and Informatics, University of Tennessee Health Science Center
Systems genetics combined with in a rapid fine mapping strategy in a reduced complexity cross identifies Rgs7 and other candidates underlying opioid addiction traits
Opioid addiction is a heritable substance use disorder, yet very little in known regarding its genetic basis. Forward genetics in mammalian model organisms can facilitate the discovery of genetic factors underlying addiction-relevant behavioral traits that could translate to humans. We recently used a reduced complexity cross (RCC) between closely related substrains of C57BL/6 to identify Cyfip2 as a genetic factor underlying binge eating. Here, we performed a systems genetic analysis of behavioral responses to the mu opioid receptor agonist oxycodone (OXY) and the antagonist naloxone (NAL) in this RCC. We identified a single major quantitative trait locus (QTL) on distal chromosome 1 that selectively influenced OXY-induced locomotor activity and spontaneous withdrawal in the elevated plus maze (160-187 Mb; Figure 1). Mendelian inheritance in the RCC obviates the need for generating congenics on an isogenic background. Thus, we focused solely on the genotype within the QTL interval and devised a strategy to rapidly fine map the locus by backcrossing and phenotyping selectively chosen F2 mice containing recombination events within the QTL interval. Notably, we reduced the size of the QTL interval by more than 10 Mb within a single generation. The QTL region captures a neurobehavioral QTL hotspot (172-178 Mb) that we are currently further dissecting. Cis-eQTL analysis of striatal tissue from a subset of 23 OXY-tested F2 mice identified three positional candidate genes, including Rgs7, Aim2, and Cadm3. Interestingly, Rgs7 was the only gene whose expression levels significantly correlated with OXY behaviors (r=0.38-0.53), further implicating this gene as a high priority candidate for validation via gene editing. A secondary genome-wide differential gene expression analysis of the QTL interval within the same OXY-treated mice confirmed these results and identified a gene network associated with two major hub genes normally associated with neurodegeneration – amyloid precursor protein and microtubule-associated protein tau. We also identified QTLs, eQTLs, and candidate genes associated with naloxone-induced, aversion-related freezing (Onecut2) as well as methamphetamine-induced locomotor activity (Gabra2). To summarize, systems genetics and fine mapping in the RCC is an efficient strategy for rapidly identifying high confidence candidate genes and neurobiological mechanisms associated with opioid addiction traits.Figure 1. A major QTL on distal chromosome 1 influences OXY-induced locomotor activity. F2 mice were trained acutely and chronically with either saline (SAL; i.p.) or OXY (1.25 mg/kg; panels A-C; 20 mg/kg/5 days; panels D-F) and assayed for locomotor activity and anxiety-like behavior in the elevated plus maze (EPM) 16 h after the final injection for spontaneous opioid withdrawal. A genome-wide QTL scan identified a major QTL on chromosome1 influencing the acute locomotor response to OXY as well as spontaneous OXY withdrawal in the EPM. For both behaviors, the J allele was associated with a greater OXY response that was specific and not observed with SAL.

Rodrigo Gularte Mérida1, Carole Charlier1 and Michel Georges1,2
1Unit of Animal Genomics, GIGA–Research
2Faculty of Veterinary Medicine, Université de Liège, 4000 Liège, Belgium
Little evidence for transgenerational genetic effects in the trascriptome of isogenic derived mice
Experiments carried out in C. elegans support the existence of paternal transgenerational genetic effects[1], i.e. the effect of untransmitted paternal alleles on the offspring’s phenotype. To test whether such effects might also occur in mammals, we generated multiple C57BL/6J (B6) isogenic cohorts mice derived from several (C57BL/6J x A/J consomic) F1 sires harboring an A/J chromosome (MMU 15, 17, 19 or X) in an otherwise B6 background— and B6 purebred females. In total we developed three distinct N2 backcrosses and one N3 backcross generating a total of 833 male mice apparently isogenic B6 mice. Global gene expression via RNA-seq in 50 pooled libraries from five tissues showed 53 genes significant differential expression between isogenic derived and B6 controls. Of these 10 were found differentially expressed in a technical replication via RT-qPCR by measuring individual gene expression in the 200 sequenced males. Further analysis of these 10 genes in 800 additional biological replicates from N2 littermates, N2 mice from an independent backcross, and N3 backcross mice showed differential expression in Mid1, Crem, and Gm26448. The expression of Mid1 was caused by a strain specific duplication event on the XY-Pseudo-Autosomal Region on MMU Y. In contrast the genes Crem and Gm26448 have no evidence of strain specific variants and showed a 0.63x higher (p < 0.0001) and -0.34x lower (p = 0.009) expression, respectively. Finally, Crem and Gm26448 were measured again in 17 additional samples from their respective strains and >17 matched controls, where Gm26448 showed a -0.7x (N2 littermate) and -0.4x (N2 independent backcross) under-expression but both failed to achieve statistical significance in the other cohorts. We have systematically explored the hypothesis paternal transgenerational genetic effects in the mouse. The series of experimental backcrosses failed to provide sufficient evidence to show a paternal genetic contribution to the offspring’s phenotype in a consistent manner. Thus, despite that transgenerational effects have been shown to exist in C. elegans knockout lines, in the mouse, under natural allelic variation, this phenomenon appears to be extremely rare, and practically undetectable with current technologies.Gregory W Carter, Catrina Spruce, Anna L Tyler, Robyn L Ball, Wendy Pitman, Narayanan Raghupathy, Michael Walker, Gary A Churchill, Kenneth Paigen, Petko M Petkov
The Jackson Laboratory, Bar Harbor, ME, 04609, USA
Genetic variation modifies epigenetic states that mediate steroid response and gene expression QTL in mice
Epigenetic changes such as histone modifications and DNA methylation can modulate transcript expression through chromatin state reorganization. Natural genetic variation potentially alters these modifications, providing a molecular mechanism for many of the abundant cis-acting gene expression quantitative trait loci (eQTL) that associate local genetic variation with differences in transcript levels. Importantly, such variation may mediate the individual response to drug treatments. However, the effects of genetic variation on regulatory regions and the extent to which alterations in DNA methylation and local chromatin configuration play a role in them have not been extensively studied. We use purified hepatocytes from a set of nine inbred mouse lines, including the founders of the Collaborative Cross and DBA/2J, to study how genetic variation affects baseline epigenetic marks as well as the response to the glucocorticoid dexamethasone. By combining RNA-seq, bisulfite DNA-seq, and ChIP-seq assays for H3K4me1, H3K4me3, H3K27me3, and H3K27ac, we obtained a detailed view of the impact of genetic variation on both epigenetic marks and transcript abundances. We found that strain-specific variation in both gene expression and epigenetic marks is abundant, and is much greater than the consequences of dexamethasone treatment in terms of both number of loci affected and the magnitude of effects. Furthermore, the effect of treatment varies substantially across mouse strains, both at the genetic and epigenetic level, providing evidence of coordinated variation in molecular responses to a common immunosuppressant. Finally, our results demonstrate local eQTL detected in mouse intercross populations such as the Diversity Outbred and BxD may be broadly due to cis-acting genetic variation in promoter and enhancer activities.David L. Aylor
Center for Human Health and the Environment & Bioinformatics Research Center, North Carolina State University, Raleigh, NC
Genetic and environmental control of gene regulation: eQTL, epigenetics, and GxE interactions
Genetics and the environment each play important roles in shaping individual molecular biology, but it has been difficult to separate the relative contributions of each factor. New population-based mouse resources such as the Collaborative Cross (CC) now enable experiments that will elucidate the effects of genes and environmental exposures with unprecedented power. We hypothesize that polymorphic regulatory elements drive gene expression differences between individuals and are crucial components of environmental response. For example, our initial results show that genetic variants drive susceptibility to diethylstilbestrol (DES), and result in abnormal reproductive tract development, infertility, and uterine gene expression changes. Mouse models of DES have been well studied, but the genetic component of DES response remained uncharacterized until our current screen in the CC. A substantial number of genes are differentially expressed in the uterus between DES-exposed and control mice in both susceptible and resistant strains, but only 61% of these genes are shared. We hypothesize that the best candidate genes will be affected by both strain and treatment – eQTLs that exist only in a specific environmental context. In separate experiments, we have established that open chromatin profiles vary widely due to genetic background. In fact, over 25% of peaks in a DNase hypersensitivity assay were absent in at least one strain. Epigenomic profiling has the potential to identify many local eQTL directly and improve our ability to fine map eQTL. Lastly, we have performed meta-analyses of two expression quantitative trait loci (eQTL) experiments done in similar populations under two different environmental conditions. Our results suggest distinct sets of tissue-specific versus environment-specific eQTL. These approaches help us to better identify and interpret eQTL, and they will ultimately help elucidate the connections between genetics, epigenetics, exposures, and phenotypes.Amelie Baud, Francesco Paolo Casale, Oliver Stegle
European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton, Cambridgeshire, CB10 1SD, United Kingdom.
Using genetics to understand the influence of the social environment.
We know that people who live together influence each other. However, quantifying the effect of the social environment on any given phenotype is difficult and only the most obvious mechanisms of social influence have been investigated (e.g. ‘phenotypic contagion’, see below).
We recently showed that the phenotype of one individual can be affected by the genetic makeup of its social partners (social genetic effects[1], SGE). We now show that SGE can be used to dissect the mechanisms whereby one individual influences another, interacting individual.
Our analyses used two large datasets collected on CFW outbred mice[2,3] and focused on social influences between cage mates. We first investigated the extent to which variation in one phenotype arises from variation in the same phenotype of the cage mates (“phenotypic contagion”). We calculated the genetic correlation between direct and social effects (where direct effects refer to effects of a mouse’s genes and environment on its own phenotype), and mapped both direct and social effects so as to compare direct and social QTLs. We show that phenotypic contagion is not sufficient to explain social effects and that more complex scenarios need to be investigated. To do so and uncover potentially novel traits of cage mates that affect phenotypes of interest, we identified specific genes whose variation in one mouse generates phenotypic variation in the mouse’s cage mates. For example, we identified Growth Factor Receptor Bound Protein 10 (Grb10) as a gene that controls stress levels of cage mates. Additional information on Grb10, specifically an observation of barbering by Grb10 mouse mutants[4], suggests that aggressive grooming mediates the SGE detected. We stress that allogrooming was not measured in the CFW datasets, so that this putative mechanism was identified purely from genetic associations (SGE).
Our findings shed light on a new dimension of genetics and illustrate how a hypothesis-free, genetics-driven approach can be used to study the influence of the social environment. A better understanding of social effects will contribute to improving general welfare and medicine.
Allison T. Knoll1, Kevin Jiang2and Pat Levitt1,2
1Program in Developmental Neurogenetics, Institute for the Developing Mind, The Saban Research Institute, Children’s Hospital Los Angeles, Los Angeles, California 90027, USA
2Department of Pediatrics, Keck School of Medicine of the University of Southern California, Los Angeles, California 90089, USA
Understanding heterogeneity in social behavior using QTL mapping in BXD mouse strains
Humans exhibit broad heterogeneity in affiliative social behavior. Twin and family studies demonstrate that individual differences in core dimensions of social behavior are heritable, yet there are knowledge gaps in understanding the underlying genetic and neurobiological mechanisms. Heterogeneity of social behavior also is evident in neurodevelopmental disorders, even in individuals with the same causal mutation. Such heterogeneity increases the complexity of developing more effective clinical treatments. We hypothesize that the genes that cause variation in typical social behavior act in concert with disorder-related genes to influence clinical heterogeneity. Using the BXD genetic reference panel (GRP) of mice, we measured four domains of affiliative social behavior—social approach, social recognition, direct social interaction (partner sniffing), and vocal communication—in two behavioral tasks that are used routinely by neuroscientists. Behavioral phenotyping of 50 strains demonstrated continuous variation in social and non-social measures that occur during the tasks. There was moderate to high heritability of social approach sniff preference (0.31), ultrasonic vocalization (USV) count (0.39), direct partner sniffing (0.51), locomotor activity (0.54-0.66), and anxiety-like behavior (0.36). Principle component analysis identified 5 independent factors that underlie variation in social and non-social traits. Significant quantitative trait loci for USV count on Chr 18 and locomotor activity on Chr X were identified by genome wide mapping, with suggestive loci for all traits with one surprising exception—partner sniffing in the DSI task. The statistically significant loci contained putative quantitative trait genes (QTGs), including a number of promising candidates based on their reported functions. Several key findings emerged from this initial analysis. First, the BXD GRP exhibits heritable variation in sociability that is independent of variation in activity and anxiety-like phenotypes. Second, the results provide new evidence that direct interaction with a social partner— a highly heritable and ethological domain of affiliative sociability—is highly polygenic. The data provide a foundation for the mapping and the identification of functional natural variants, which will provide a new understanding of variation in typical social development and contributions to neurodevelopmental disorder risk.Sarah M. Neuner1, Kristen M.S. O’Connell2, Catherine C. Kaczorowski1,2
1University of Tennessee Health Science Center, Memphis, TN, 371163, USA
2The Jackson Laboratory, Bar Harbor, ME, 04609, USA
Discovering Genetic Modifiers of Alzheimer's Disease Using Novel Mouse Models
Background: Recent studies of familial AD cases suggest genetic factors can delay the onset and progression of cognitive deficits by decades. Therefore, these genetic factors may provide key targets for treatment and prevention of AD. However, significant barriers restrict our ability to discover the genetic mechanisms of resilience using conventional human studies. Resilient individuals are often asymptomatic and thus escape the attention of medical geneticists. Animal models of AD provide a powerful resource for longitudinal and in-depth analyses, but have traditionally been studied on one genetic background. These limitations create a critical need for innovative approaches that take advantage of ‘high-risk’ FAD mutations in animal models in combination with genetic diversity in order to identify resilient genetic backgrounds that can be used to gain a better understanding of molecular mechanisms underlying resilience.
Methods: To discover genetic modifiers of FAD, we generated a novel mouse panel of genetically diverse mice carrying FAD mutations in APP and PS1. Mice were phenotyped across the lifespan using construct valid cognitive and neuropathological assays, and genetic interval mapping was used to identify loci associated with resilience to cognitive decline and Aβ pathology.
Results: We observed wide variation in the age at onset and severity of AD symptoms in our FAD mouse population, which parallels variation observed in humans with high-risk FAD mutations. Results from our genetic interval mapping identified multiple genomic regions associated with resilience or susceptibility to AD, including the APOE locus and several novel quantitative trait loci.
Conclusions: As our goal is to discover novel genes and molecular networks that govern human AD resilience, genes and molecular networks underlying resilience to AD in our mouse studies will be integrated with existing human genetic data to prioritize candidates with high translational relevance. High-priority genes associated with resilience to AD in human cohorts will be targeted via genome-editing in relevant mouse models for functional validation. Since clinical and pathological hallmarks of FAD parallel those of sporadic late-onset AD cases, we expect modifiers discovered in our mouse studies will broadly generalize to all forms of AD.
Tamara Martin, Stuart Nicklin, Delyth Graham, Martin W McBride
BHF Glasgow Cardiovascular Research Centre, University of Glasgow, Glasgow, UK, G12 8TA
Osteopontin over-expression increases H9c2 cell size
Background. The stroke-prone spontaneously hypertensive rat (SHRSP) is an excellent model of human cardiovascular disease and develops increased left ventricular mass index (LVMI) prior to the onset of hypertension. We identified a quantitative trait locus (QTL) for LVMI on chromosome 14. Using chromosome 14 congenic strains and gene profiling, have identified osteopontin (Spp1) as a positional candidate gene. The aim of this study is to validate Spp1 as a functional candidate gene affecting cell size.
Methods. H9c2 cells were seeded (at a seeding density of 3.4x104 cells/ well) 24hrs prior to transfection with Spp1 cloned into pcDNA1 (5ug) for 48hrs, using Xfect transfection reagent. Conditioned media was obtained from cells post-transfection and incubated with fresh H9c2 cells for 48hrs. Extracellular vesicles were isolated from conditioned media (CM) via ultracentrifugation, verified by NanoSight, re-suspended in PBS and placed onto fresh H9c2 cells for 48hrs. EVOS (crystal violet) microscopy images were analysed using ImageJ software. Cell data was analysed using ANOVA with Dunnett’s post hoc test.
Results. H9c2 cells transfected with Spp1 cDNA derived from the SHRSP exhibited a significant increase in cell size compared with cells transfected with pcDNA vector (pcDNA 107.9±1.4 vs SHRSP 141.8±2.3, p<0.001). Significantly increased Spp1 mRNA expression was confirmed in transfected cells by qRT-PCR (RQ Cells 1.0±0.09, pcDNA1 2.0±0.31, SHRSP Spp1 cDNA 58.2±4.55*, *p<0.001 compared to cells only). Conditioned media taken from SHRSP transfected cells produced a significant increase in H9c2 size compared with pcDNA vector controls (pcDNA 67.9±1.1 vs SHRSP Spp1 133.0±2.9, p<0.001). EVs isolated from conditioned media transfected with SHRSP significantly increased H9c2 cell size compared to pcDNA vector controls (pcDNA 96.6±1.5 vs SHRSP Spp1 152.9±2.6, p< 0.001).
Discusssion Collectively these data validate Spp1 as a functional candidate gene affecting cell size and suggest that over-expression of Spp1 promotes an increase in cell size via extracellular vesicle signalling. Further studies are required to characterise extracellular vesicle content and downstream mechanisms leading to hypertrophy.
Jeremy W. Prokop1, Nan C. Yeo2, Christian Ottmann3, Emily J. Ross1, Chris McDermott-Roe4, Aron M. Geurts4, Brian A. Link5, Eric M. Mendenhall1, Jozef Lazar1, Howard J. Jacob1
1HudsonAlpha Institute for Biotechnology, Huntsville, Alabama 35806, USA.
2Department of Genetics, Harvard Medical School, Boston, Massachusetts, 02115, USA.
3Department of Biomedical Engineering and Institute for Complex Molecular Systems, Eindhoven University of Technology, Eindhoven, Netherlands.
4Human and Molecular Genetics Center, Medical College of Wisconsin, Milwaukee, Wisconsin 53226, USA.
5Department of Cell Biology, Neurobiology and Anatomy, Medical College of Wisconsin, Milwaukee, Wisconsin 53226, USA.
Characterization of coding and noncoding variants for SHROOM3 and chronic kidney disease from rat to human.
Interpreting genetic variants is one of the greatest challenges impeding the analysis of the rapidly increasing volume of genomic data from patients. For example, SHROOM3 is a recently associated risk gene for chronic kidney disease (CKD), yet the causative genetic mechanism(s) that alter risk through the SHROOM3 allele are unknown. We developed an analytical pipeline that integrates genetic, computational, biochemical, CRISPR/Cas9 modification, molecular, and physiological data to characterize coding and non-coding genomic variants in the rat and human genome, and applied this workflow to the SHROOM3 risk locus for CKD. In the rat, SHROOM3 is one of several CKD risk alleles that have been combined together to minimally recapitulate the multifactorial kidney phenotype of the FHH rat strain, with initial RNAseq providing disease biomarkers and mechanistic insight into CKD progression. Using our strategy for the human SHROOM3 allele, we identified a novel SHROOM3 transcriptional start site, which results in a shorter isoform lacking the PDZ domain, and is regulated by a common non-coding upstream allele associated with CKD. This variant disrupts binding of TCF7L2 and FOXO1 in podocyte cells and the single variant (rs17319721) alters transcription levels of SHROOM3 as determined by variant insertion using CRISPR/Cas9. A full assessment of the ExAC database for >60,000 sequenced human exomes revealed several rare coding variants in SHROOM3 that are likely to alter function, including a coding variant (P1244L) we have associated with CKD. Using our analytical pipeline, we determined that P1244L attenuates the interaction of SHROOM3 with 14-3-3 binding and alters signaling through the Hippo pathway, a known mediator of CKD. These data demonstrate multiple new SHROOM3-dependent genetic and molecular mechanisms that regulate CKD and demonstrate a novel analytical pipeline with broad application to systematically characterizing risk mechanisms of CKD and other complex diseases.Laura J Lambert1, Anil K Challa1, Michal Mrug2, Darwin Bell2, Bradley Yoder3, Robert A Kesterson1
1Departments of Genetics
2Medicine and
3Cell Biology, University of Alabama at Birmingham, Birmingham, Alabama, 35233, USA
Generation of new rat models of ciliopathies
Ciliopathies affect nearly every organ system and are defined by genetic mutations resulting in compromised structure or function of cilia. Cilia defects cause a multitude of phenotypes in humans including cystic kidney, liver, and pancreatic diseases as well as blindness, deafness, hydrocephalus, congenital heart malformations, infertility, obesity, diabetes, and cognitive defects. Mutations in multiple genes have been identified in ciliopathy patients with some occurring at high frequencies.
Animal models for the ciliopathies are important for analyzing candidate cilia regulated signaling pathways and contribute greatly to our fundamental knowledge about ciliogenesis and cilia function. While mice have been the primary mammalian model for ciliopathy research, largely due to advantages in genetic engineering, recent advances with nuclease-based genome editing approaches, such as CRISPR-Cas9, now afford researchers the ability to use rat models. Mouse models of ciliary function have produced key basic science discoveries; however, this mammalian species has limitations in translating to human disease. Unfortunately, many obstacles prevent laboratories from using rats as in vivo ciliopathy models including time, effort, cost, and the expertise needed to generate models using the new engineering approaches.
To facilitate the development, distribution and use of genetically modified rat models with broad utility to analyze cilia sensory and signaling activity, pathophysiology of cilia disorders, and for preclinical assessment of potential therapeutic targets, we have recently generated multiple novel rat models using CRISPR-Cas9 and transgenic approaches. Polycystin-2 (Pkd2), a ciliary localized Ca2+ channel, was disrupted using nucleases targeted to two exons. While mice heterozygous for Pkd2 do not develop cysts, all Pkd2 heterozygous rats develop cysts in both kidneys. Thus, our rat models more closely mimic the dominant cystic disease present in autosomal dominant polycystic kidney disease (ADPKD) patients. Additionally, a null allele of IFT88 has been created, and establishment of a conditional knockout allele is underway, with germline transmission of a 3’ loxp allele to be further modified to complete the conditional state.
To accelerate in vivo analyses of ciliopathies and efficacy of potential therapeutics, we are also establishing biosensor lines to evaluate cilia and related pathways, including lines to assess changes in Ca2+ and cAMP levels. Recent preliminary data shows integration of an R-GECO ciliary Ca2+ sensor into the rat genome, with germline transmission of the allele established and functional studies forthcoming. To enhance basic and translational research into ciliopathies using rats, we will continue to establish conditional, congenital, and human disease allele models with mutations in ciliopathy genes.
Ashley C. Johnson1, Michael R. Garrett1,2
1Pharmacology and Toxicology, University of Mississippi Medical Center
2Medicine (Nephrology) and Pediatrics (Genetics), University of Mississippi Medical Center
Identification of novel genetic factors involved in altered kidney development in the HSRA congenital solitary kidney rat
The HSRA rat is a unique polygenic model of congenital abnormalities of the kidney and urogenital tract [1]. In particular, 50-75% of offspring develop with a single kidney. The single kidney (HSRA-S) exhibits nephron deficiency (~20% less nephrons than normal kidney), glomerular and kidney hypertrophy, and elevated single nephron GFR, which leads to progressive kidney injury and decline in kidney function with age [2]. HSRA-S rats are also highly sensitive to hypertension-induced kidney injury [3]. While the physiological implications of the HSRA model have been well-studied, it remains unknown the specific genetic factors that cause failure of one kidney to develop as well as the mechanism of altered development/nephron deficiency in the solitary kidney. A recent genome-wide association study (GWAS) performed by others using the heterogeneous stock rat (progenitor of the HSRA model), identified genomic loci on chromosomes (RNO) 4, 10, and 14 associated with the solitary kidney trait. The confidence interval for the RNO4 locus is ~4.2 Mb containing 14 genes, the RNO10 interval is ~1.5Mb containing 15 genes, and the RNO14 locus is consistent with region linked to solitary kidney in the ACI rat. The whole genome of the HSRA rat was sequenced to a depth of 18X coverage using the Illumina NextSeq500 platform. Sequence alignment and annotation identified 1,776,635 SNP, 314,786 INDEL, and 25,346 structural variants. RNA sequencing of embryonic kidney isolated at key stages of nephrogenesis (E14.5, 15.5, and 16.5) between single kidney and two-kidney control identified dysregulation of genes linked to cellular movement (p-value= 2.36E-06 - 1.49E-02 n=78/451), cell morphology (p-value= 1.86E-05 - 1.49E-02 n=82/451) and cell-to-cell signaling and interaction (p-value= 8.38E-05 - 1.49E-02 n=68/451). Based on the GWAS, whole genome sequencing, and RNA sequencing, the causative genomic loci were refined to specific genes [c-Kit (RNO14), Fam58b and Nlk (RNO10), and Met and Wnt2 (RNO4)] that could play a role in the solitary kidney trait and nephron deficiency in the HSRA model. In summary, the identification of the specific gene defect, modifier genes, and signaling pathways that lead to loss of one kidney and nephron deficiency will provide a mechanistic link between the solitary kidney genotype and the development of kidney dysfunction exhibited by the HSRA rat.Sarah Galla1, Harshal Waghulde1, Xi Cheng1, Saroj Chakraborty1, Blair Mell1, Venkatesha Basrur2, Bina Joe1,
1Physiological Genomics Laboratory, Center for Hypertension and Personalized Medicine, Department of Physiology and Pharmacology, University of Toledo College of Medicine and Life Sciences, Toledo, OH
2Department of Pathology, University of Michigan, Ann Arbor, MI.
Quantitative proteomic analysis of endothelial cells in G-protein coupled estrogen receptor (Gper1) knockout rats
G-protein coupled estrogen receptor (Gper1), a previously identified orphan receptor, now known to be a receptor for estrogen and aldosterone, has been localized to blood vessels, heart, and the central nervous system. Gper1 knockout models have been developed in mice and rats and have shown both increased and decreased blood pressure as compared to controls. In addition, a Gper1 agonist, G1, caused decreased blood pressure when injected into mice. Women with a GPERP16L amino acid substitution in their GPER1 protein have increased BP. In order to study the effects of this receptor further, we created a novel CRISPR/Cas9 gene deletion model of the Gper1 gene on a Dahl Salt-Sensitive (S) rat background. Hemodynamic and vascular phenotypic studies demonstrated that the Gper1 knockout rats had lower blood pressure and improved vascular function compared to the S rat in an endothelial dependent manner. As this improved vascular function was observed in an endothelial dependent manner, we hypothesize that deletion of Gper1 causes quantitative alterations in the cellular proteome of endothelial cells. To test this hypothesis and explore mechanisms behind the observed BP lowering effect in Gper1 knockout rats, a quantitative proteomic analysis was conducted on endothelial cells isolated and cultured from the thoracic aorta of Gper1 knockout rats and wildtype S rats. Endothelial cell cultures were digested and quantitatively analyzed by mass spectrometry. A total of 101 statistically significant differentially expressed proteins were identified, of which 97 proteins were with higher expression in the Gper1 knockout rats. We prioritized the most significantly upregulated protein, Cd99, as a potential mechanistic link downstream of the functionality of Gper1 in endothelial cells. Cd99 is a transmembrane protein involved in both permeability and transendothelial migration. We found increased transendothelial migration of macrophages in the Gper1 knockout rats compared to the wild-type S rats in vitro. These results coupled with the improved vascular function and lowered BP of Gper1 knockout rats lead us to believe that macrophages have a protective role in the Gper1 knockout ratsSaroj Chakraborty1, Ying Nie1, Xi Cheng1, Blair Mell1, Sarah Galla1, Peter J Czernik2, Beata Lecka-Czernik2, and Bina Joe1
1Physiological Genomics Laboratory, Center for Hypertension and Personalized Medicine
2Center for Diabetes and Endocrine Research, Department of Physiology and Pharmacology, University of Toledo College of Medicine, Toledo, OH.
A polymorphic variant of Secreted Phosphoprotein 2 as a quantitative trait nucleotide linked to the heritability of blood pressure and bone mineral density in a gender dependent manner
Hypertension is a complex polygenic disease caused by a combination of inherited (genetic) and environmental factors. Rat models of inherited hypertension serve as tools to dissect and prioritize genetic factors as candidate genes causing hypertension. One such candidate gene prioritized through systematic linkage and substitution mapping is Spp2 or Secreted Phosphoprotein 2. A single non-synonymous G/T polymorphism between the Dahl Salt-Sensitive (S) rats and Spontaneously Hypertensive Rats (SHR) at the Spp2 locus was hypothesized to cause a reduction in blood pressure (BP) and bone mineral density (BMD) observed in the S.SHR congenic strain spanning the Spp2 locus. To test this hypothesis, a novel rat model was generated using the CRISPR/Cas9 precision-engineering technology, whereby the ‘G’ allele at the Spp2 locus of the S rat was replaced by the ‘T’ allele of the SHR rat. Protein modeling prediction by SWISSPROT indicated a significantly altered protein structure of the Spp2 protein in the resultant Spp2 knock-in rescue model. Following transgenesis, pups born were genotyped and grouped into founders and non-founders. The founder and non-founder rats were isogenic except for the Spp2 locus, wherein the founder S rats contained the ‘G’ allele and the non-founder S rats contained the ‘T’ allele. Both male and female (founder and non-founder) rats were fed a high salt (2% NaCl) containing diet and their BP was monitored by radiotelemetry. Systolic BP of the Spp2 knock-in male rats was significantly lower compared to that of the non-founder S rats. However, systolic BP of the Spp2 knock-in female rats was significantly higher compared to that of the non-founder S rats. Again, trabecular bone of this high salt fed male and female rats were collected and analyzed by micro-CT. Although, the bone volume by total volume ratio is not significantly different in male Spp2 knock-in, but the bone volume by total volume ratio was significantly lower in female Spp2 knock-in rats. Therefore, only Spp2 knock-in female rats were showing lower bone mineral density. These data provide conclusive evidence for a single nucleotide polymorphism within the Spp2 gene as a quantitative trait nucleotide (QTN) responsible for the inheritance of blood pressure and bone mineral density.Funding for this work to BJ from the NHLBI/NIH (HL020176) is gratefully acknowledged.
Lauren B Shunkwiler1, Cody C Ashy2, Royal M Pipaliya2, Benjamine Van Peel1, Jan Guz3, Michael J Kern3, Yang Zhao1Bart MG Smits1
1Department of Pathology and Laboratory Medicine, Medical University of South Carolina, Charleston, SC 29403, USA
2College of Charleston, Charleston, SC, 29403, USA
3Department of Regenerative Medicine, Medical University of South Carolina, Charleston, SC 29403, USA
CRISPR-Cas9 Targeting of 16q12.1 Breast Cancer Susceptibility Locus to Generate Allelic Series of Rat Mutants Results in Altered Tox3 Expression
As the incidence of breast cancer diagnoses continues to surge, it is imperative to understand mechanisms that underlie susceptibility to this disease. Genome-wide association studies (GWAS) have identified regulatory variants that influence likelihood of developing breast cancer. The human chromosomal locus 16q12.1 attracts attention for it contains GWAS-identified variants that uniquely associate with breast cancer risk in populations of European and African American women. The correlated polymorphisms comprising these variants exist in non-coding regions of this locus, providing the possibility that these mutations alter gene expression. The most likely candidate gene, TOX3, encodes a transcription factor that has previously been found to regulate Estrogen Receptor (ER)-responsive genes in breast cancer cells. The risk allele of the variant strongly associated with ER+ breast cancer in women of European descent is associated with lower expression levels of TOX3. We hypothesize that non-coding breast cancer-associated polymorphisms on 16q12.1 regulate TOX3, subsequently modifying risk of developing ER+ breast cancer.
To begin exploring TOX3 as a candidate breast cancer susceptibility gene, we used CRISPR-Cas9 technology to genetically engineer an allelic series of mutations in the rat genome. The rat is the preferred rodent model for ER+ breast cancer. Rats were generated with deletions or knock-in mutations in the portion of the Tox3 locus orthologous to the human risk-associated region. Functional studies are possible with these new rat models because we obtain viable mutants across all genotypes, despite partial embryonic lethality in homozygous Tox3 knockouts.
The allelic series displays variable levels of Tox3 downregulation in the mammary gland, suggesting there are multiple Tox3-regulatory elements in this non-coding region. Mutants showing altered Tox3 expression also show significant effects on mammary gland development, namely on ductal elongation and the numbers of terminal end buds and branch points. The phenotypes implicate Tox3 in mammary stem/progenitor cell biology potentially through ERα gene regulation. Interestingly, Tox3 knockout rats also present a morbid obesity phenotype and previous publications demonstrate the role of TOX3 in neuronal tissue, suggesting possible pleiotropic effects of mutations in the TOX3 locus.
Ongoing studies are focused on the functional role of Tox3 in mammary gland biology and carcinogenesis to elucidate the mechanism of susceptibility to ER+ breast cancer. Understanding breast cancer risk will ultimately lead to innovative strategies and therapeutic approaches aimed at preventing the development of breast cancer.
Isha S. Dhande1, Mykola Mamenko2, Sterling Kneedler1, Yaming Zhu1, Oleh Pochynyuk2, Scott E. Wenderfer3, Michael C. Braun3, Peter A. Doris1,
1Institute of Molecular Medicine, University of Texas Health Science Center at Houston, Houston, TX 77030
2Department of Integrative Biology and Pharmacology, University of Texas Health Science Center at Houston, Houston, TX 77030
3Department of Pediatrics, Baylor College of Medicine, Houston, TX 77030
A natural mutation in Stim1 creates a major defect in immune function in stroke-prone spontaneously hypertensive rats
The genetic mechanism of hypertensive end organ injury may involve variation in genes participating in inflammation and its regulation. We identified a novel truncating mutation in the hypertensive end organ injury-prone spontaneously hypertensive rat (SHR-A3/SHRSP) line affecting the C-terminus of STIM1, a protein involved in the store-operated Ca2+ entry (SOCE) pathway. SOCE is required by T cells to activate the transcription factor NFAT and regulate T cell proliferation and cytokine production. T cell receptor (TCR) stimulation depletes intracellular Ca2+ stores, activates the ER Ca2+-sensor STIM1 and results in SOCE and is essential for transcriptional control of cellular immune responses. The expression of STIM1 was significantly reduced in SHR-A3 lymphocytes when compared with the injury-resistant SHR-B2 strain, which expresses the ‘wild-type’ STIM1. In CD4+ T cells, SOCE triggered by thapsigargin as well as T cell receptor (TCR) activation was dramatically reduced in SHR-A3, but not in SHR-B2. Flow cytometric analysis of circulating T cell subsets revealed comparable levels of CD4+ and CD8+ T cells in both lines, however circulating CD4+CD25+FoxP3+ Tregs were reduced in SHR-A3 compared to SHR-B2 (4.34±0.59 vs 7.12±0.33%, p=0.01). T cell cytokine production in response to TCR stimulation was markedly impaired CD4+ T cells from SHR-A3 compared with SHR-B2 (IL-2: 168 ±83.4 vs 1385±377.0 pg/mL, p=0.01; IFNγ: 235±69.5 vs 2119±434.7 pg/mL, p=0.002). IL-2 and IFNγ production was completely inhibited by Pyr6, an inhibitor of STIM1-dependent SOCE. Further, TCR-induced proliferation and activation-induced apoptosis were both impaired in CD4+ T cells from SHR-A3 when compared with SHR-B2. Based on our findings, we conclude that TCR-mediated effector signaling is impaired due to defective SOCE in SHR-A3 rats. Defects in SOCE in SHR-A3 attributable to STIM1 mutation alter T cell function, reduce Treg numbers and may disturb regulatory interactions between T cells and other immune cells involved in end organ injury.Olivia L Sabik1,2, Gina M Calabrese2, Cheryl L Ackert-Bicknell3, Charles R Farber1,4
1Center for Public Health Genomics, School of Medicine, University of Virginia, Charlottesville, VA 22908
2Department of Biochemistry and Molecular Genetics, School of Medicine, University of Virginia, Charlottesville, VA 22908
3Center for Musculoskeletal Research, University of Rochester Medical Center, University of Rochester, Rochester, NY 14624
4Department of Public Health Sciences, University of Virginia, Charlottesville, VA 22908
Systems genetics identifies novel genes and gene networks influencing osteoblast activity
Osteoporosis is a disease characterized by low bone mineral density (BMD) and increased risk for fracture. Identifying novel modulators of bone formation remains a goal in the pursuit of safe, effective therapies that promote bone anabolism. While many of the critical signaling pathways impacting bone formation by the osteoblast are known, our knowledge is incomplete, and it is unclear how osteoblast activity is regulated at a systems-level. BMD is a “snap shot in time” measure—it is the sum of bone formation and resorption and cannot be used as surrogate phenotype for bone formation alone. Similarly, serum markers of bone formation can be confounded by other physiologic and pathologic factors from a genetics point of view. In this study, we used an innovative systems genetic approach to identify novel genes and gene networks modulating osteoblast activity in the Collaborative Cross (CC). We isolated calvarial osteoblasts from 48 CC strains, differentiated them into mature, mineralizing osteoblasts, and measured their levels of in vitro mineralization. Our measure of in vitro mineralization was highly heritable (H2=0.52; p = 1.4 x 10-52) in the CC. Additionally, we observed a positive correlation between in vitro mineralization and cortical bone area/total area (BA/TA) of the femur (0.43, p = 0.008), supporting the physiological relevance of in vitro osteoblast activity. To identify genetic drivers of in vitro mineralization we performed a genetic analysis in the CC and identified one quantitative trait locus (QTL) on Chromosome 10. Calvarial osteoblasts from CC strains homozygous for the CAST/EiJ (CAST) haplotype in the QTL region displayed lower levels of mineralization. To determine the gene responsible for the association we identified colocalizing local expression QTL (eQTL). Cytoskeleton Associated Protein 4 (Ckap4) was the only gene whose expression was significantly associated with the CAST haplotype, which increased Ckap4 expression in osteoblasts. Ckap4 is a known receptor of WNT antagonist Dickkopf1 (Dkk1), in the context of cancer, and WNT signaling plays a role in the regulation of bone formation. We also constructed a weighted gene co-expression network using transcriptomic data on the osteoblasts from 42 CC strains and identified three modules of genes associated with in vitro mineralization. We are currently prioritizing hub genes from these modules for mechanistic follow up. This work has provided us with new insight into the molecular mechanisms that regulate osteoblast activity.Basel Al-Barghouthi1, and Charles R. Farber1,2
1Department of Biochemistry and Molecular Genetics and Center for Public Health Genomics, School of Medicine, University of Virginia, Charlottesville, VA 22908
2Department of Public Health Sciences, School of Medicine, University of Virginia, Charlottesville, VA 22908
Identification of genes affecting bone strength-related traits in Diversity Outbred mice.
Osteoporosis is a disease characterized by reduced bone strength and an increased risk of fracture. Approximately 40 million individuals in the U.S. alone suffer from, or are at a high risk for developing, osteoporosis. Bone strength-related traits are highly heritable, but bone strength cannot be directly measured in humans. Instead bone mineral density (BMD), a clinically accessible trait associated with fractures, has historically been used for genetic studies of osteoporosis in humans. However, BMD only explains part of the variation in bone strength, limiting the development of a comprehensive understanding of osteoporosis. In order to fill this knowledge gap, we are performing a comprehensive genetic analysis of bone strength using Diversity Outbred (DO) mice. Currently, we are phenotyping a population of 800 DO mice to identify quantitative trait loci (QTL) for over 60 bone-strength related traits, including bone morphology and microarchitecture, bone remodeling markers, marrow adiposity, in vivo bone dynamics and biomechanics. In addition to the comprehensive suite of clinical phenotypes, we are also generating RNA-seq data on purified cortical bone and cultured osteoblasts and osteoclasts (N=192/sample type). These data will be used for eQTL based fine-mapping and the generation of co-expression and Bayesian networks to identify modules of genes associated with clinical phenotypes. We have currently phenotyped over 250 mice and generated RNA-seq profiles on 96 cortical bone samples. Our novel and innovative approach to gene discovery has the potential to significantly increase our understanding of the basic biological processes that underlie variation in bone strength, leading to the discovery of novel therapeutic targets for the prevention and treatment of bone fragility.Chie Naruse1,2, Shinwa Shibata2, Masaru Tamura3, Takayuki Kawaguchi2, Kanae Abe2, PI1, Kazushi Sugihara1,2, Tomoaki Kato4, Takumi Nishiuchi4, Shigeharu Wakana3, Masahito Ikawa5, Masahide Asano1,2
1Institute of Laboratory Animals, Graduate School of Medicine, Kyoto University, Kyoto, Japan.
2Division of Transgenic Animal Science, Advanced Science Research Center, Kanazawa University, Kanazawa, Japan.
3Technology and Development Team for Mouse Phenotype Analysis, RIKEN BioResource Center, Tsukuba, Japan.
4Division of Functional Genomics, Advanced Science Research Center, Kanazawa University, Kanazawa, Japan.
5Animal Resource Center for Infectious Diseases, Research Institute for Microbial Diseases, Osaka University, Suita, Japan.
New insights on the role of Jmjd3 and Utx in axial skeletal formation in mice
Jmjd3 and Utx are demethylases specific for lysine 27 of histone H3 (H3K27). Previous reports indicate that Jmjd3 is essential for differentiation of various cell types such as macrophages and epidermal cells in mice, whereas Utx is involved in cancer and developmental diseases in humans and mice, and Hox regulation in zebrafish and nematodes. Here, we report that Jmjd3, but not Utx, is involved in axial skeletal formation in mice. A Jmjd3 mutant embryo (Jmjd3−/−), but not a catalytically inactive Utx truncation mutant (Utx-/y), showed anterior homeotic transformation. Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) and microarray analyses showed reduced Hox expression in both Jmjd3−/− embryos and tailbuds, suggesting that Jmjd3 plays a regulatory role in Hox expression during axial patterning. Chromatin immunoprecipitation analyses of embryo tailbud tissue showed trimethylated H3K27 (H3K27me3) to be at higher levels at the Hox loci in Jmjd3−/− mutants compared to wild-type tailbuds. Demethylase-inactive Jmjd3 mutant embryos showed the same phenotype as Jmjd3−/− mice. These results suggest that the demethylase activity of Jmjd3, but not that of Utx, affects mouse axial patterning in concert with alterations in Hox gene expression.Aysar Nashef1, Ervin Weiss2, Yael Houri-Haddad1, Fuad A. Iraqi3
1Department of Prosthodontics, Dental school, The Hebrew University, Hadassah Jerusalem, Israel
2School of Dental Medicine, Tel-Aviv University, Tel-Aviv, Israel
3Department of Clinical Microbiology and Immunology, Faculty of Medicine, Tel-Aviv University, Tel-Aviv, Israel
Transcriptome analysis reveals candidate genes affecting susceptibility to periodontitis in the Collaborative Cross (CC) mouse population
Background: Periodontitis is a common inflammatory disease characterized by alveolar bone loss. To characterize the global transcriptome of periodontitis and suggest candidate genes associated with the disease we applied massively parallel RNA-seq of samples from susceptible and resistant CC lines to alveolar bone loss phenotype.
Material and Methods: Different CC lines were screened for their alveolar bone loss susceptibility phenotype induced by periodontal infection using µCT technique. RNA samples of 24 hemi maxillae from two susceptible lines, two resistant lines and two “over resistant” lines (4 mice per line, 2 infected and 2 control) were extracted and sequenced on a HiSeq platform. Normalization and differential expression of data were performed with the DESeq2 package (version 1.12.4) and visualized in R. The significance threshold for all comparisons was taken as padj<0.1. Biological processes that are up- or down-regulated in connection with susceptible mice were identified using the annotation tool DAVID and related processes clustered using the functional annotation clustering tool.
Results: 1142 genes were significantly up-regulated in susceptible lines and related to immune-response, NF-kappa B-signaling and immunodeficiency pathways. 1197 genes were significantly down regulated and connected to cell differentiation, ATP binding proteins. Deferentially expressed genes were subsequently interrogated with fine-mapped QTL analysis associated with periodontitis susceptibility in CC population. Eight genes were underlie the mapped QTL and differentially expressed and suggested to be strong candidate genes associated with periodontitis.
Conclusion: Our results demonstrate the power of the CC population in detecting susceptibly genes to periodontitis through combined RNA-seq and QTL analysis.
Michael-John G. Beltejar1, Jun Zhang1,2, Dana A. Godfrey1, Olivia Hart3, Renata Rydzik3, Douglas J. Adams3, Cheryl L. Ackert-Bicknell1
1Center for Musculoskeletal Research, Univ. of Rochester Medical Center, Rochester, NY
2Department of Orthopedics, Zhejiang Provincial People’s Hospital, Zhejiang, China
3Department of Orthopaedic Surgery, UConn Health, Farmington, CT
Genetic variation influences bone matrix composition resulting in varied femoral strength among inbred mice.
Osteoporosis is a complex disease characterized by progressive reduction in bone quantity and alterations in bone matrix quality. This often culminates in fractures that significantly alter quality of life and increase mortality risk. Although bone mineral density (BMD) is the current basis for surveillance, diagnosis, and therapy, it only captures 50% of fracture resistance because it minimally reflects contributions from bone shape and composition. Understanding the contributions of composition is necessary to develop novel and more comprehensive screening and treatment. The goal of this work was to determine how defined genetic variations in bone matrix quality results in altered bone strength. To accomplish this, we leveraged the genetic diversity of 7 inbred mouse strains. Three-point bending was used to measure flexural strength of femurs from 20-week old male mice. Moments of inertia for diaphyseal cross sections of femurs were measured using microCT. Tissue modulus and strength were calculated using beam theory. Areal BMD of the whole body was measured using DXA. Finally, matrix composition of humeri was assessed using Fourier-Transform Infrared Spectroscopy (FTIR). Differences among strains were determined by one-way ANOVA with a Student’s T post hoc test with a Bonferroni correction. All of these determinants were highly reproducible within a strain, yet differed among strains, suggesting high heritability for these phenotypes. Multiple regression using parameters of composition (BMD, mineral: matrix, carbonate:phosphate, crystallinity) and geometric properties (I and I/c about ML axis) confirmed that whole-bone mechanical strength is predominantly determined by I/c, crystallinity and the mineral to matrix ratio (min:matrix). Tissue modulus is predominantly determined by crystallinity and min:matrix. Poignantly, contributions of aBMD to whole-bone strength is redundant. We then identified the main determinants of strength for each strain. For example, despite having lower BMD and geometrically smaller bones than B6, CAST femora have greater tissue strength, tissue modulus, and a mineral to matrix ratio, resulting in greater structural strength than B6 mice. This suggests that genetically regulated matrix properties might be involved in offsetting deficiencies of size and shape to produce femurs of similar structural strength. These genetic differences can now be exploited to identify the molecular pathways involved in governing structural bone strength.Lauren J. Donoghue1,2, Alessandra Livraghi-Butrico3, Kathryn M. McFadden1, Joseph M. Thomas1, Gang Chen3, Charles R. Esther Jr3,4, Barbara R. Grubb3, Wanda K. O’Neal3, Richard C. Boucher1, and Samir N. P. Kelada1-3
1Department of Genetics, University of North Carolina, Chapel Hill, NC
2Curriculum in Genetics and Molecular Biology, University of North Carolina, Chapel Hill, NC
3Marsico Lung Institute, University of North Carolina, Chapel Hill, NC
4Pediatric Pulmonology, University of North Carolina, Chapel Hill, NC
Identification of trans protein QTL for secreted airway mucins and a causal role for Bpifb1
Background Mucus hyper-secretion is a hallmark feature of asthma and other muco-obstructive airway diseases. The two secreted mucins, MUC5AC and MUC5B, represent the major glycoprotein components of mucus and contribute to its biophysical properties. We performed an unbiased, genome-wide search to identify quantitative trait loci (QTL) in mice that control the levels of MUC5AC and MUC5B protein secreted into the airways after allergen sensitization and challenge.
Methods We applied a house dust mite allergen challenge model of asthma to both founder lines (n=8) and incipient lines (n=154) of the Collaborative Cross (CC). We collected bronchoalveolar lavage (BAL) samples 72 hours after allergen challenge and quantified MUC5AC and MUC5B by agarose gel electrophoresis followed by Western blotting. Each CC mouse was genotyped on a high-density Affymetrix array and gene expression was also measured by arrays.
Results CC founder lines sensitized and challenged with allergen exhibited statistically significant differences in both MUC5AC and MUC5B, providing evidence of heritability. Incipient CC lines exhibited a broad range of MUC5AC and MUC5B in BAL. QTL mapping identified distinct, trans protein QTL loci on Chr 13 for MUC5AC (at 75 Mb) and Chr 2 for MUC5B (at 154 Mb), respectively. Statistical analysis of the MUC5B QTL allele effects allowed us to narrow the Chr 2 QTL region to a ~2 Mb region, and we identified Bpifb1 (bactericidal/permeability-increasing protein fold containing family B, member 1) as a candidate gene. We show that BPIFB1 is expressed in the large airways and colocalizes with MUC5B, and that its expression parallels Muc5b mRNA and protein levels after allergen challenge. Finally, we found that MUC5B expression is upregulated in Bpifb1 knockout (vs. wildtype) mice after allergen challenge and that this difference also affects the critical pulmonary host defense parameter of mucociliary clearance.
Conclusion Our results indicate that the concentrations of MUC5AC and MUC5B in BAL after allergen sensitization and challenge are controlled by distinct, trans-acting genetic loci. Variation in the gene Bpifb1 is causally related to MUC5B levels and mucociliary clearance. Based on additional analysis of gene expression data, we speculate that protein-protein interactions underlie the observed relationship between MUC5B and BPIFB1, and we have identified a candidate causal variant in Bpifb1 to investigate mechanistically in future studies.
Beverly A Richards-Smith, Judith Blake, Carol Bult, Anna Anagnostopoulos, Susan Bello, Michelle Knowlton, Monica McAndrews, Hiroaki Onda, Jill M Recla, Deborah Reed, Monika Tomczuk, Laurens Wilming, Cynthia L Smith
Mouse Genome Informatics (MGI), The Jackson Laboratory, Bar Harbor ME 04609-1500, USA
What's in a name? Standardized nomenclature for mouse and rat
Meaningful scientific communication is founded on shared understanding by all participants of the terminology employed. This is best achieved by use of a mutually accepted lexicon. Different biological disciplines may use different terms for the same object or concept and the same term for different objects, and lab jargon and lay language can further distort meaning. The lexicon of genetics is designed unequivocally to identify genes and other functional genomic features, their mutant and variant forms, and genetically defined strains of experimental organisms.
The human and mouse genomes contain ~25,000-30,000 protein coding genes and an expanding assortment of known functional RNAs, regulatory elements, and highly evolutionarily conserved non-coding sequences of largely unknown function. The nomenclature must evolve to incorporate newly discovered genomic entities, new classes of engineered genetic/genomic modifications enabled by novel technologies, e.g., mitochondrial replacement and CRISPR/Cas, and different types of rodent strains/stocks, such as the mouse Collaborative Cross lines and their derived recombinant inbred strains.
Authoritative nomenclature guidelines are established by species nomenclature committees, such as the International Committee on Standardized Nomenclature for Mice, the Rat Genome and Nomenclature Committee, and the HUGO (Human Genome Organization) Gene Nomenclature Committee. These committees coordinate the nomenclature of orthologous genes among different species. The guidelines are posted on individual species Web sites that include searchable databases where users can find approved designations for specific genes, mutations and variants, etc.
The recently established Alliance of Genome Resources (AGR; http://www.alliancegenome.org/) seeks to integrate data from the Gene Ontology (GO) Consortium (http://www.geneontology.org/), the Mouse Genome Database (MGD; http://www.informatics.jax.org/), the Rat Genome Database (RGD; http://rgd.mcw.edu/wg/home), FlyBase, Saccharomyces Genome Database (SGD), WormBase, and the Zebrafish Information Network (ZFIN) to further promote these model organisms’ utility in interrogating the genetic/genomic context of human biology, health and disease. The AGR database is an integrated resource for gene orthology, nomenclature, ontology, disease relationships, and gene function and expression profiles, accessible from a unified web interface. Standardized nomenclature will assist in consistent searching and retrieving all gene, variant and strain information across species in this resource.
We will demonstrate standardized nomenclature for various classes of mouse and rat strains; genetic variants including alleles at quantitative trait loci (QTL); and mutant alleles including classically targeted and endonuclease-mediated mutations. Official Nomenclature Guidelines for mouse and rat genes, alleles, chromosomal aberrations and strains are accessible from the Nomenclature Home Pages at MGI (http://www.informatics.jax.org/mgihome/nomen/index.shtml) and RGD (http://rgd.mcw.edu/nomen/nomen.shtml).
Supported by NIH grant HG000330
Michal Pravenec1, Vaclav Zidek1, Vladimir Landa1, Petr Mlejnek1, Jan Silhavy1, Tomas Mracek1, Josef Houstek1, Hynek Strnad2, Ludmila Kazdova3, Harry Smith4, Laura Saba4, Boris Tabakoff4
1Institute of Physiology, Czech Academy of Sciences, Prague, Czech Republic
2Institute of Molecular Genetics, Czech Academy of Sciences, Prague, Czech Republic
3Institute for Clinical and Experimental Medicine, Prague, Czech Republic
4Department of Pharmaceutical Sciences, University of Colorado School of Pharmacy, Aurora, USA
Genetic dissection of brown adipose tissue function in rat recombinant inbred strains
Recently, brown adipose tissue (BAT) was suggested to play an important role in lipid and glucose metabolism in rodents and possibly also in humans. In the current study, we used quantitative trait locus (QTL), genomic sequence and RNA expression analyses in the BXH/HXB recombinant inbred (RI) strains, derived from Brown Norway (BN) and spontaneously hypertensive rats (SHR), to identify some of the genetic determinants of BAT function and its role in the pathogenesis of metabolic disturbances. Linkage analyses revealed significant QTL associated with interscapular BAT mass on chromosome 4 and two closely linked QTL associated with glucose oxidation and glucose incorporation into BAT lipids on chromosome 2. Using quantitative transcriptional data and weighted gene co-expression network analysis (WGCNA), we identified 1,147 gene co-expression modules in the RNA extracted from BAT of the BXH/HXB panel. The “Coral14.1” co-expression module includes the hub gene Cd36 which plays an important role in fatty acid transport and triglyceride utilization in BAT. The eigengene QTL for the “Coral14.1” module overlaps with the QTL for interscapular fat mass on chromosome 4, and the module eigengene values are significantly correlated with the fat mass across the RI strains. The “Darkseagreen” module eigengene QTL overlaps the QTL associated with glucose oxidation and BAT lipid synthesis on chromosome 2, and this module’s eigengene values are significantly correlated with these biochemical parameters across the RI strains. The “Darkseagreen” module has the flavin-containing monooxygenase 5 (Fmo5) and sortilin 1 (Sort1) as the most connected transcripts. The functions of these genes can be clearly related to the associated phenotypes. We also searched the areas delineated by the phenotypic QTLs for functional polymorphisms. Cd36 harbors a deletion variant in the SHR. SHR-Cd36 transgenic rats with wild type Cd36, when compared to SHR, had reduced BAT mass. We also identified Wars2 (tryptophanyl tRNA synthetase 2 (mitochondrial)) as a positional candidate which is dysfunctional in the SHR. SHR-chr.2 congenic and SHR-Wars2 transgenic rats, with wild type Wars2 gene, showed increased glucose oxidation and incorporation into BAT lipids when compared to SHR. BAT mass, glucose oxidation and incorporation into BAT lipids significantly correlated with metabolic and hemodynamic parameters in RI strains. In summary, our results demonstrate an important role of both differences in transcriptional levels and functional genetic polymorphisms in regulating BAT mass and function and consequently lipid and glucose metabolism. Supported in part by Czech Science Foundation (13-04420S) to MP and NIH/NIAAA (R24AA013162) to BT.Kathie Sun1,2, Greg Keele1,2, Daniel Pomp1, and William Valdar1,3
1Department of Genetics, University of North Carolina at Chapel Hill, NC, USA
2Curriculum in Bioinformatics and Computational Biology, University of North Carolina at Chapel Hill, NC, USA
3Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, NC, USA
Estimating heritability and genetic correlation of exercise-related traits in Diversity Outbred mice.
Narrow sense heritability is a fundamental concept in quantitative genetics that describes the proportion of phenotypic variation for a given trait that is due to additive genetic variation within the population. In addition, many complex traits are highly polygenic and thus the result of variation in many common alleles acting in concert. Shared causal loci (pleiotropy) results in correlations between traits. Genetic correlation is an estimate of the portion due to additive genetic effects common to pairs of traits. These two quantities are often estimated using likelihood-based methods, which do not easily produce interval estimates in variance components. In addition, default implicitly uniform priors on variance components in maximum likelihood estimation may be informative and influence estimation. We used phenotype and genotype data from experiments in two groups of Diversity Outbred mice, a structured model population, to derive and implement a Bayesian procedure for estimating heritability and genetic correlation. Subsequently, we used these procedures to obtain point and interval estimates from complex traits, including exercise- and metabolism-related metrics. Heritability and genetic correlation estimates have been published in previous mouse studies for several of the traits explored in this project. Overall, our results for traits related to running and body size broadly agree with the literature, particularly estimates from the group of mice fed with a consistent diet.Williams, Evan G1, Jha, Pooja2, Roy, Suheeta3, Ingels, Jesse3, Wang, Xu2, Hasan, Moaraj1, Zamboni, Nicola1, Lu, Lu3, Auwerx, Johan2, Williams, Robert W3, Aebersold, Ruedi1
1Department of Biology, Institute of Molecular Systems Biology, ETH Zurich, Zurich CH-8093, Switzerland
2Laboratory of Integrative and Systems Physiology, École Polytechnique Fédérale de Lausanne, Lausanne CH-1015, Switzerland
3Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Center, Memphis, TN 38163, United States
Metabolism in the Aging Liver: Gene-by-Environment Interactions Across the BXD Population
The most prevalent complex metabolic diseases are driven by a wide array of genetic and environmental factors which are compounded by the passage of time. The BXD genetic reference population is a cohort of ~150 distinct inbred mouse strains derived from C57BL/6J and DBA/2J which we have previously to have highly variable metabolic phenotypes. In this study, we have followed 2210 individuals from 86 BXD strains on both low-fat and high-fat diets across their lifespans (~300 to ~1200 days). 659 of these individuals were taken for tissue collection at 7, 12, 18, and—when possible—24 months of age to provide tissues for time course analysis of the aging process. We are now analyzing the livers from these individuals for a multi-omic analysis including the transcriptome, proteome, and metabolome to observe how metabolic pathways are regulated across time, genetic background, and diet. These data are then merged with phenotypic information collected in the same individuals (e.g. clinical blood parameters and body weight over time) and compared against historical phenotype and omics records collected in the same BXD strains under similar dietary and housing conditions. This systems biology approach is being mined for de novo hypothesis discovery for subsequent mechanistic studies on the etiology of aging and the development of metabolic diseases.Shweta Ramdas1, Ayse Bilge Ozel2, Yu-yu Ren3, Ying Ma4, Nathan R. Qi5, Mary K. Treutelaar6, Lauren G. Koch6, Steven L. Britton6, PI1, Charles F. Burant5, Jun Z. Li1,2
1Department of Computational Medicine and Bioinformatics, University of Michigan Ann Arbor, Michigan 48104 USA
2Department of Human Genetics, University of Michigan Ann Arbor, Michigan 48104 USA
3Department of Psychiatry, University of California San Diego, 92093-0667 USA
4Department of Biostatistics, University of Michigan Ann Arbor, Michigan 48104 USA
5Department of Internal Medicine, University of Michigan Ann Arbor, Michigan 48104 USA
6Department of Anesthesiology, University of Michigan Ann Arbor, Michigan 48104 USA
CONVERGENT GENOMIC ANALYSES OF A RAT MODEL OF AEROBIC CAPACITY AND METABOLIC HEALTH
A decrease in aerobic exercise capacity (AEC) in humans is associated with increased risks of many diseases, including obesity, insulin resistance, hypertension, and type 2 diabetes. To understand the biological underpinnings of AEC, we previously established two rat lines by divergent selection of intrinsic endurance running capacity. After 32 generations, the high capacity runners (HCR) and low capacity runners (LCR) differed by ~9-fold in running distance, and diverged in body fat, blood glucose, and other health indicators. In this study we crossed HCRs and LCRs to generate >490 F2 animals and genotyped them on a genomewide panel of ~700K SNPs. We also collected RNA-Seq data for skeletal muscle samples to identify eQTLs. The QTL for running distance and the eQTLs are being combined with other results, including signatures of positive selection and known variants from founder strains to identify likely causal variants for AEC. We also collected metabolomics data for a subset of the animals, and in a separate cohort analyzed the impact of aging and acute exercise on gene expression patterns in the skeletal muscle. We found that transcripts for mitochondrial function are expressed higher in HCRs than LCRs at both rest and exhaustion and for both old and young animals. Expression of cell adhesion and extracellular matrix genes tend to decrease with age. This and other age effects are more prominent in LCRs than HCRs, suggesting that HCRs have a slower aging process and this may be partly due to their better metabolic health. By combining genetic and genomic approaches we expect that functional alleles at multiple loci will be implicated. Many of these may be directly relevant for the corresponding human phenotypes or, at a minimum, provide clues to important pathways that could be targeted for improving human metabolic health.Anna L. Tyler, Bo Ji, Daniel M. Gatti, Steven C. Munger, Gary A. Churchill, Karen L. Svenson, Gregory W. Carter
The Jackson Laboratory, Bar Harbor, ME, 04609, USA
Epistatic networks jointly influence phenotypes related to metabolic disease and gene expression in Diversity Outbred mice
Genetic studies analyzing multiple traits simultaneously have the potential to create multi-scale models of complex traits that link genetic variation, gene expression, and physiological data. Incorporating epistasis, or genetic interactions, into these studies has the further potential to identify specific mechanistic interactions underlying high-level physiological traits. In this study, we modeled epistasis influencing body composition, serum biomarkers, and liver transcript abundances in a population of 474 Diversity Outbred (DO) mice. The DO mouse population offers advanced genetic diversity and phenotypic variability, as well as high genetic mapping resolution, and is well suited to the dissection of complex trait genetic architecture. Using weighted gene co-expression network analysis (WGCNA), we condensed transcript data into summary phenotypes with enriched biological processes. We then used combined analysis of pleiotropy and epistasis (CAPE) to model epistasis that simultaneously influenced the module phenotypes, body composition, and serum biomarkers. We identified a network of epistatic interactions between alleles of different ancestries and saw evidence of both genetic synergy and redundancy between haplotypes. A subset of loci showed sex-specific effects. We also identified a number of alleles that potentially protect individuals from the effects of a high-fat diet. Although the epistatic interactions explained small amounts of trait variance, they frequently resulted in individuals with extreme phenotypes, particularly when interacting alleles were derived from different parental backgrounds. Furthermore, we were able to use epistatic interactions as context for generating hypotheses about the roles of specific genes in complex traits. Our approach demonstrates that going beyond the single-locus to single-trait map to model multiple loci and multiple traits simultaneously creates information-rich genetic maps of complex traits that can be further dissected to generate gene-level hypotheses.Youjie Zhang1, Blair Mell1, Xi Cheng1, Steven L. Britton2, Lauren G. Koch2, and Bina Joe1
1Center for Hypertension and Personalized Medicine, Department of Physiology and Pharmacology University of Toledo College of Medicine and Life Sciences, Toledo, OH
2Department of Anesthesiology, University of Michigan, Ann Arbor, MI.
Gut microbiotal dysbiosis and increased risk for complex polygenic diseases emerge with genomic selection for low aerobic exercise capacity
Bi-directional host-microbiotal interactions are increasingly being recognized as important factors contributing to the pathophysiology of complex polygenic diseases. While host genomes are inherited, the microbiome is acquired as a result of the composition of the microbiota that choose to reside within the host. The identities of the factors that determine which microbes choose to reside in a given host are unknown. Given the evidence for host-microbiotal interactions in health and disease, we hypothesized that host genomes exert a selection pressure to influence the types of microbiota that reside in the host and that the resultant microbiotal composition is a pivotal determinant of the divide between health and disease of the host. To test this hypothesis, we developed models of health and disease by inbreeding rats divergent in aerobic exercise capacity. These novel inbred strains were developed by >20 generations of brother-sister mating of the selectively-bred low aerobic capacity rats and high aerobic capacity rats and named as LCR/BJ and HCR/BJ, respectively. Inbred HCR/BJ rats had significantly higher aerobic exercise capacity than inbred LCR/BJ rats as recorded by their total running distance to exhaustion of 1151 m vs. 130 m, respectively, p< 0.001. Next-generation sequencing of the whole genomes of inbred HCR/BJ and LCR/BJ rats revealed considerable differences in their genomic sequences. To test whether this selection for differential genomic sequences resulted in any alterations in microbiota, fecal microbial communities of inbred LCR/BJ and HCR/BJ rats were profiled using 16S rRNA sequences. Interestingly, principal coordinates analysis displayed distinct clustering of the two cohorts (ANOSIM analysis, weighted UniFrac, P = 0.002), indicating significant phylogenetic differences of the microbial community structures between inbred LCR/BJ and HCR/BJ rats. Firmicutes/Bacteriodetes ratio was higher in the LCR/BJ rats compared with HCR/BJ rats suggesting the possibility of the increased risk for diseases in the LCR/BJ rats. Compared with inbred HCR/BJ rats, inbred LCR/BJ rats exhibited elevated blood pressure, increased body weight, lower circulating mean corpuscular hemoglobin, and higher neutrophil-to-lymphocyte ratio. Metabolic and behavioral phenotyping is currently underway. Overall, these studies provide clear evidence for the ability of the host genome to shape the microbiome and identify an inverse co-evolutionary relationship between host genomic factors conferring enhanced aerobic exercise capacity and microbiotal dysbiosis driving disease risks.Paul L Maurizio1, Martin T. Ferris1, Gregory R. Keele1, Darla R. Miller1, Alan C. Whitmore1, Ande West1,2, Clayton R. Morrison1, Kelsey E. Noll1,3, Kenneth S. Plante1, Adam S. Cockrell2, David W. Threadgill4, Fernando Pardo-Manuel de Villena1,5, Ralph S. Baric2, Mark T. Heise1,5 and William Valdar1,5
1Department of Genetics, UNC-Chapel Hill, Chapel Hill, NC, 27514, USA
2Department of Epidemiology, UNC-Chapel Hill, Chapel Hill, NC, 27514, USA
3Department of Microbiology & Immunology, UNC-Chapel Hill, Chapel Hill, NC, 27514, USA
4Institute of Genome Sciences and Society, Texas A&M University, College Station, Texas 77433-2470
5Lineberger Comprehensive Cancer Center, UNC-Chapel Hill, Chapel Hill, NC, 27514, USA
Diallel analysis reveals Mx1-dependent and independent effects driving influenza virus severity
Influenza A virus (IAV) is a respiratory pathogen that causes substantial morbidity and mortality during both seasonal and pandemic outbreaks. Infection outcomes in unexposed populations are affected by genetics, but the host genetic architecture is not well understood. Here we obtain a broad view of how heritable factors affect a mouse model of host response to IAV infection using an 8×8 diallel of the eight inbred founder strains of the Collaborative Cross (CC). Expanding on a prior statistical framework for modeling treatment response in diallels, we explored how additive genetics, dominance, parent-of-origin effects, epistasis, and sex-specific versions thereof modify acute host response to IAV through 4 days post-infection. Heritable effects in aggregate explained about 60% of the variance in IAV-induced weight loss, with much of this attributable to a pattern of additive effects that becomes more prominent through day 4 post-infection and that is consistent with previous reports of anti-influenza Mx1 polymorphisms segregating between these strains. The pattern of effects after controlling for Mx1 was less distinct, distributed between several heritable categories, but nonetheless accounted for about 34% of the remaining variance. The observed additive effects largely recapitulate haplotype effects observed at the Mx1 locus in a previous study of the incipient CC (preCC); we show they also replicate in a new CC recombinant intercross (CC-RIX). Furthermore, the subspecies origin of functional Mx1 alleles determines their level of dominance when combined with null Mx1 alleles. This has implications for mapping studies using heterozygous individuals such as recombinant F1s and outbred mice.Sarah M. Neuner1,2, Shengyuan Ding2, Catherine C. Kaczorowski2
1Neuroscience Institute, University of Tennessee Health Science Center, Memphis, TN, 372163, USA
2The Jackson Laboratory, Bar Harbor, ME, 04609 USA
Hp1bp3 influences neuronal excitability and cognitive function
Recently, heterochromatin protein 1 binding protein 3 (Hp1bp3) was identified as a novel regulator of cognitive aging using a combination of forward genetics and a global knock-out mouse model. Through gene set enrichment analyses, we discovered that genes whose hippocampal expression correlated with that of Hp1bp3 were significantly enriched for terms related to plasma membrane localization and for functions related to neuronal excitability. As changes in hippocampal neuronal excitability and plasticity have been posited to underlie learning and memory processes, we hypothesized Hp1bp3 may be mediating its effects on cognitive aging via the regulation of hippocampal neuronal excitability. To test this hypothesis, we performed a targeted intrahippocampal knockdown of Hp1bp3 in adult mice, followed by behavioral analyses and slice electrophysiology. Mice receiving shRNA for Hp1bp3 exhibited both working and contextual fear memory deficits, demonstrating that decrease of Hp1bp3 in the hippocampus is sufficient to induce cognitive deficits reminiscent of those observed in global knock-out mice. In addition, these mice exhibited decreased hippocampal neuronal excitability as indicated by an increase in the slow after-hyperpolarization (sAHP), providing a candidate mechanism for Hp1bp3-mediated changes in cognitive function. As Hp1bp3 is known to regulate chromatin accessibility and gene expression, it is likely that Hp1bp3 regulates the expression of critical ion channels and receptors in the plasma membrane. Ongoing work in the lab will examine this question in more detail. Finally, hippocampal sAHP plasticity in disrupted in mouse models of Alzheimer’s disease, suggesting Hp1bp3 may play a role in AD-related cognitive deficits, a possibility which will be investigated in future studies.Sumana R. Chintalapudi1, Doaa N. Maria1,3, XiangDi Wang1, Janey L. Wiggs5, Robert R. Williams1,2,4, Monica M. Jablonski1,2,4
1Department of Ophthalmology, The Hamilton Eye Institute, The University of Tennessee Health Science Center, Memphis, TN, USA
2Department of Anatomy and Neurobiology, The University of Tennessee Health Science Center, Memphis, TN, USA
3Department of Pharmaceutical Sciences, The University of Tennessee Health Science Center, Memphis, TN, USA
4Department of Genetics, Genomics and Informatics, The University of Tennessee Health Science Center, Memphis, TN, USA
5Department of Ophthalmology, Harvard Medical School, and Massachusetts Eye and Ear Infirmary, Boston, Massachusetts, USA
Cacna2d1: a novel therapeutic target for lowering IOP.
Glaucoma is a leading cause of blindness worldwide and intraocular pressure (IOP) is the only modifiable risk factor. Genetic variability is a major contributor to interpersonal differences in responses to IOP lowering therapies. In this study, we combined systems genetics using the aged BXD mice strains with human GWAS and pharmacology to define and validate a genetic modifier of IOP associated with individual genetic variations.
Methods: IOP was measured in 66 BXD strains at 10-13 months using a TonoLab. Genomic regions modulating IOP were identified using Quantitative Trait Locus (QTL) analyses. Stringent refinement based on QTL mapping, correlation analyses and single nucleotide polymorphisms (SNPs) was performed to identify candidate genes. Subcellular localization of candidates in mouse and human eyes was determined by immunohistochemistry. The GLAUGEN/NEIGHBOR consortium database was used to identify SNPs within the candidate genes associated with glaucoma in humans. IOP lowering effects of nimodipine and pregabalin were evaluated as eye drops in C57BL/6J (B6), BXD14 (B parent allele) and BXD48 (D parent allele) strains (n=6 for each strain). The minimum effective concentration was determined.
Results: A single eQTL on Chr 5 was identified with a significant likelihood ratio statistic (LRS) of 19.6. Cacna2d1 was identified as a cis-regulated candidate gene. CACNA2D1 is expressed in ciliary body, trabecular meshwork, retina and optic nerve in mouse and human eyes. GLAUGEN/NEIGHBOR POAG meta-analyses revealed a SNP (rs4732474) nominally associated with POAG (P=0.001009). This candidate is a component of an L-type voltage gated calcium channel (CACN) regulating ionic transport. Nimodipine⎯an antagonist for the CACN pore⎯reduced IOP in B6 by 18.4±1.7%, BXD14 by 31.3±3.5% and BXD48 by 3.2±2.0% compared to baseline. Similarly, pregabalin, an antagonist specific for the α2δ1 subunit of CACN, reduced the IOP in B6 by 20.4±5.5%, BXD14 by 28.5±3.5% and BXD48 by 14.2±3.9%.
Conclusions: This is the first study to combine systems genetics, bidirectional studies using human GWAS, and pharmacology to identify and validate a genetic modulator of IOP. Both nimodipine and pregabalin lowered IOP significantly in strains with the B parent allele compared to strains with D parent allele. In the future, similar pharmaco-genetic studies could pave the way for improved POAG therapies tailored to individual genotypes.
Danny Arends1, Stefan Kärst1, Sebastian Heise1, Gudrun A. Brockmann1
1Humboldt-Universität zu Berlin, Invalidenstraße 42, D-10115 Berlin, Germany
Transmission distortion and genetic incompatibility of alleles in mice predisposed for obesity
While direct additive and dominance effects on obesity have been investigated repeatedly using association tests, additional genetic factors contributing to this complex trait have been scarcely investigated. To assess genetic background effects we tested the transmission frequency of alleles using genotype data from three generations of mice from our advanced intercross (AIL) population (C57BL/6NCrl x Berlin Fat Mouse Inbred).
Allele transmission ratio distortions (TRD) were investigated using allele transmission asymmetry tests. Detection of genetic incompatibilities using SNP chip data is often impractical due to the large amount of pairwise tests leading to a multiple testing issues, we reduced the number of tests only looking for incompatibilities between identified TRD.
In total we detected 85 regions showing significant TRD, in particular in close proximity to the centromere. In TRD regions, genes are located that point to genetic determinants in the immune system contributing to obesity. Genes in paternally biased regions showed pathway overrepresentation for fatty acid degradation and PPAR signaling; maternally biased regions harbored genes associated with olfactory transduction, Wnt signaling and nitrogen metabolism. Interestingly, by using Genenetwork we found that 23 of these TRD regions have previously been associated with obesity in BXD mice.
Using DNA-Seq data we observed genes in TRD regions contained 6.5 times as many non-synonymous SNPs compared to genes in randomly selected regions (182 vs. 28.7 ± 8.1). Testing for genetic incompatibilities between TRD regions identified 56 pairs showing signatures of genetic incompatibilities (P < 0.01). Further investigation of these regions could provide new targets for investigating genetic adaptation and determinants of obesity.
Felix L. Struebing, Eldon E. Geisert
Department of Ophthalmology, Emory University, Atlanta GA
Regulatory element networks underlying QTLs and disease loci: Towards a better understanding of non-coding variation in complex traits
One of the surprising discoveries of genome-wide association studies is that 95% of loci associated with disease are located outside protein coding exons [1]. Simultaneously, the functional importance and tissue-specificity of many of these non-coding regulatory elements was addressed by large research consortia such as RIKEN or ENCODE. The association of phenotype and genotype, however, remains hard to predict for complex diseases, in which an unknown number of genomic loci might interact to form a disease trait. Therefore, new methodological approaches are needed to advance knowledge in this field.
To approach this issue, we present a computational strategy that integrates data from arbitrary sources and is capable of predicting regulatory elements such as promoters, enhancers, genomic anchor sites and their respective transcription factor binding sites (TFBS), in a tissue-specific manner. Adding genotype information to this data allows construction of regulatory element networks that could help to better understand the origin of complex traits or diseases. To simplify this task, we designed an interactive application suite written in R/Shiny that incorporates algorithms from many other R/Bioconductor packages and performs higher-level functions such as plotting or statistical analysis.
As a proof of principle, we integrated retinal transcriptome data from C57BL/6J (B6) and DBA/2J (D2) mice with publically available DNase-Seq data [2] and TFBS predictions for CTCF, a prominent genomic anchor protein functioning as insulator and loop-promoting factor between regulatory elements [3]. We found that SNPs in the 19bp long DNA-binding motif for CTCF result in a higher probability of transcripts in proximity to be differentially expressed (Figure 1). This indicates that disrupted regulatory elements result in measurable changes of RNA expression, which in turn might underlie complex traits and diseases. It also demonstrates the importance of interactions between non-coding regulatory elements, such as enhancers and promoters, in adequately controlling transcription. Future aims include extending our approach across different tissues and the BXD family of recombinant inbred mouse strains in order to comprehensively model regulatory element networks governing gene expression.
Figure 1: Average false discovery rate (FDR) profile of exons ±2Mb up/downstream of CTCF binding sites. FDR is higher when the DNA-binding motif is conserved between B6 and D2 mice (straight red line), indicating no differential expression of transcripts in vicinity. Disruption of the motif results in decreased FDR profiles encompassing a genomic area that is consistent with the size of topologically associated domains (~1Mb, dashed blue line), which are formed by CTCF loops [4]. Shaded area indicates 95% confidence interval.

Richard S. Nowakowski1, Lisa M. DiCarlo1, Kelin Xu2, Naomi Brownstein3, Jianqing Fan2,4, Cynthia Vied1
1Department of Biomedical Sciences, Florida State University, Tallahassee, FL 32312, USA
2School of Big Data, Fudan University, Shanghai, China
3Department of Behavioral and Social Medicine, Florida State University, Tallahassee, FL 32312, USA
4Departments of Statistics and Finance, Princeton University, Princeton, NJ, USA
Pattern detection in large datasets: comparison of methods to detect “switched” genes in gene expression datasets
The statistical analysis of large data sets created by genomic and proteomic analyses is a difficult and sometimes unsatisfying experience. In many cases, the appropriate statistical tests have a goal of “discovery” and for these the often used approaches of ANOVA, GLM and various clustering procedures are likely to also be the most appropriate. For some datasets and experiments, however, plausible biologically based hypotheses can be constructed. For these cases, data analysis can take a different form with specific and perhaps limited tests of hypotheses.
As a first step, we are exploring transcriptomic data from RNA-seq data for the existence of “switched” genes, which we define as genes which exhibit an “on/off” pattern of expression. We specify that “on/off” genes have only two levels, an “off” set that is indistinguishable from zero expression and an “on” state that is statistically different from the “off state and is the same (i.e., not different statistically) for all experimental conditions that are not “off”. Note that this “on/off” switched gene definition delimits only a small proportion of the genes that are differentially expressed. For example, we define “hi/lo” genes as having a non-zero low state, i.e., the low state is significantly greater than zero expression.
We have identified “on/off” switched genes in multiple data sets using two general approaches: 1) using ANOVA/GLM (with Benjamini-Hochberg FDR corrections) followed by all possible pair-wise post-hoc tests with genome wide criteria for specifying the “zero level” of expression”, and 2) clustering with various distance metrics. The ANOVA/GLM method works well with small numbers of experimental groups, especially if the number of replicates per group is reasonable; this approach also has the advantage of being exhaustive, i.e., criteria can be specified that allows all genes in the data set to be assigned to the “on/off” switched type or not. The clustering approach is the only viable option when there are large numbers of experimental groups with few replicates per group, but is also applicable the smaller groups for which the ANOVA/GLM approach can be used. We will present comparisons of these two approaches for different datasets with different numbers of experimental groups and proportions of differentially expressed genes.
Akiko Takizawa1, Nadiya Sosonkina2, Lynn Lazcares1, Jeremy W Prokop2, Teppei Goto3, Michael Grzybowski1, Aron Geurts1, Jozef Lazar2, Masumi Hirabayashi1,3, Howard J Jacob2, Melinda R Dwinell1
1Human & Molecular Genetics Center, Medical College of Wisconsin, Milwaukee, WI, 53130, USA
2HudsonAlpha Institute for Biotechnology, Huntsville, AL, 35806, USA
3National Institute for Physiological Sciences, Okazaki, Aichi, 444-8787, Japan
Generation of a rat model with a one nucleotide substitution in the MAPK1 gene to mimic mutation in a patient with undiagnosed disease
Patients with rare diseases often undergo an exhausting and expensive diagnostic odyssey. Advancements in DNA sequencing, whole exome (WES) and whole genome sequencing (WGS), offer revolutionary diagnostic tools to these patients often leading to cures and therapies. However, a considerable number of identified variants fall into category of variant of uncertain significance (VUS). To make an accurate diagnosis, the pathophysiological role of a VUS needs to be explained. Here, we report the results of the characterization of a de novo variant c.404G>C (p.R135T) in the MAPK1 gene found using WES in a female proband with the following major symptoms: microcephaly, intellectual disability, speech apraxia, ADHD, craniosynostosis, facial dysmorphic features, and cyclic vomiting syndrome. This MAPK1 missense mutation was classified as a VUS. From this result, we generated a gene modified rat model to be the first demonstration of a transgenic rat with knocked-in point mutation to mimic the patient’s variant and potential patient phenotype. The substitution of C for the wild-type G at nt 398 of the rat Mapk1 would mimic the patient mutation. The rat ES cell line (F344/NSlc-ES37/Nips, RGD ID: 10054027) was gene modified by CRISPR/Cas9 system. We obtained Mapk1 mutated rats from 8 male chimeric rats. This rat model has several phenotypes in common with the patient, including lack of weight gain and facial dysmorphic features. In parallel, we have received skin biopsies from the patient and her parents and performed cell-based assays to investigate the function of the MAPK1 variant. Interestingly, gene expression studies showed overrepresentation of mutant MAPK1 allele. In conclusion, we have evaluated different approaches to functional characterization of a VUS found in a patient with rare disease.Funded by the W.M. Keck Foundation (HJJ) and R24HL114474 (HJJ, MRD, AG).
Adrianus C.M. Boon and Traci Bricker
Department of Medicine, Washington University School of Medicine, St Louis, Missouri, 63110, USA
Genetic approach to study H5N1 influenza A virus pathogenesis
The genotype of the host has been implicated in the severity of and susceptibility to influenza A virus infections. The exact mechanism and genetic polymorphisms responsible are currently unknown. Earlier work on recombinant inbred BXD mice, derived from resistant C57BL/6 and susceptible DBA/2 mouse strains, identified three genomic loci that were associated with resistance to severe highly pathogenic H5N1 disease and mortality. More recently we tested a chromosomal substitution strain set (CSS) derived from C57BL/6 and another susceptible mouse strain, A/J, in which a single chromosome of C57BL/6J is substituted with the homologous chromosome of A/J. Of the nineteen CSS strains tested, C57BL/6J-Chr. 4 A/J was significantly more susceptible to highly pathogenic H5N1 influenza A virus than the parental C57BL/6J, indicating that genetic polymorphisms on this chromosome are responsible for the difference in H5N1. The increased susceptibility of C57BL/6-Chr. 4 A/J mice was associated with higher virus titers at days 2, 7 and 9 post infection and increased expression of IFN-b1 and type I interferon induced proteins. To help identify the genetic polymorphism and host gene on chromosome 4, we have produced a series of congenic mouse strains. Preliminary analysis suggests that the polymorphism is located between 130 and 150Mb on chromosome 4. In conclusion, genetic polymorphisms in the genome of the host can predispose to severe influenza disease and systems biology and genetics approach allows us to identify essential host proteins involved in resistance to severe influenza disease.David J. Samuelson, Saasha Le, Jennifer Sanders, and Aaron D. den Dekker.
Center for Genetics & Molecular Medicine, Department of Biochemistry & Molecular Genetics, University of Louisville School of Medicine, Louisville, Kentucky
Rat Mcs1b, Mcs3, and Mcs6 are genetic models of female breast cancer risk.
Female breast and rat mammary cancer susceptibility are complex traits with incompletely defined genetic components. Comparative genomics between these species is anticipated to increase our knowledge of genetically determined mechanisms of cancer progression and resistance. Laboratory rats (Rattus norvegicus) provide a good experimental model of female breast cancer, as rat mammary carcinomas are similar to human breast carcinomas with respect to histopathology and hormone responsiveness. Inbred rat strains differ in susceptibility to hormone-, chemical-, and oncogene-induced mammary carcinogenesis. Multiple rat Mammary carcinoma susceptibility (Mcs) and Estrogen-induced mammary cancer (Emca) QTLs have been mapped by various groups. We used independent congenic strains that defined Mcs1b, Mcs3, and Mcs6 QTLs to identify candidate susceptibility genes and variants, as well as human orthologous loci. Rat Mcs1b, located in 1.8 Mb of RNO2, is a mammary-gland cell-autonomous ortholog of the human genome-wide association-study identified female breast cancer risk locus at 5q11.2. Using genome sequence analysis and RT-QPCR we identified three Mcs1b candidate variants and three candidate genes. Rat Mcs3 was mapped to a 27.8 Mb segment of RNO1 that contains sequence orthologous to human genomic regions on multiple chromosomes, including 2, 11, and 15. These regions contain variants with reported p-values for association to breast cancer risk that are low; however, these variants did not meet a required genome-wide level of significance. Thus, based on the location of rat Mcs3, these human loci are prioritized candidates to be ultra-fine mapped and further tested in genetic association studies. Rat Mcs6 was mapped to an 8.5 Mb segment of RNO7. The orthologous human region on chromosome 12 contains variants that have met genome-wide significance levels for association with breast cancer risk. Thus, the rat genome is useful to prioritize potential risk-associated human loci for further study. And, the laboratory rat is further indicated to be useful in functional studies of genetically determined mechanisms of cancer susceptibility and resistance potentially shared by humans and rats.Liang Zhao1, Megan K Mulligan2, Lu Lu2, Robert W Williams2, Thaddeus S Nowak Jr1
1Department of Neurology, University of Tennessee Health Science Center, Memphis, Tennessee, 38117, USA
2Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Center, Memphis, Tennessee, 38117, USA
Variation in stroke vulnerability among BXD mouse strains and C57BL/6 substrains
Background Previous studies identified genetic loci impacting stroke vulnerability in mice following distal middle cerebral artery occlusion (MCAO), associated with variations in the microvasculature [1,2]. Using this model both C57BL/6J (B6) and DBA2J (D2) mice yield very small infarcts. However, combining MCAO with ipsilateral common carotid artery occlusion increases insult severity. This yields appreciable infarcts in B6 whereas brain injury in the D2 strain remains negligible. We have applied this tandem occlusion model to characterize the variation in stroke vulnerability among BXD strains, as well as among several B6 substrains.
Results Overall heritability of infarct size was 50% among mice (n=1076) of the 89 BXD strains examined, with no significant sex difference. Most strains exhibited lesions intermediate between those of the parents, but some were larger, and a third were very small like D2. Mapping identified significant loci on Chr 4 and 5, at which larger infarcts are associated with B6 and D2 alleles, respectively, and additional suggestive loci on Chr1, Chr2 and Chr4. Infarct volumes (mm3 ± SE) for J, EiJ and ByJ substrains were 11.0 ± 0.8, 10.0 ± 2.6 and 10.1 ± 0.9 (n=73, 8 and 35), whereas those for NCrl, NJ and NTac were 17.8 ± 1.0, 18.8 ± 1.3 and 17.3 ± 2.8 (n=35, 35 and 11).
Conclusions B6 and D2 mice are comparable with respect to collateral vascularization [2], so the variation in infarct size among BXD lines not surprisingly identifies novel loci impacting stroke. Expanding the data set to include new BXD lines will refine loci, strengthen correlations with other phenotypes, and ultimately suggest responsible genes and molecular networks. The almost two-fold difference in vulnerability between N and J substrains emphasizes the critical importance of background strain selection in stroke studies involving genetically modified mice. The ByJ substrain, originating from the N lineage in 1961, retains the smaller infarct phenotype characteristic of the J lineage, so the greater vulnerability of subsequently derived N substrains is due to an intervening mutation.
Leonie C. Weber1, Bernd. U. Koelsch1 and Andrea Kindler-Röhrborn1
1Instutute of Pathology, University of Duisburg-Essen, University Hospital of Essen, 45147 Essen, Germany
Variant alleles of estrogen receptor beta (Esr2) mediate sex dependence of traits to different extents
A great number of quantitative trait loci (QTL) in rodents and humans show marked sex dependence. This observation relates to physiological phenotypes as well as to disease susceptibilities. We performed two genome wide association studies to detect QTLs influencing the risk of developing chemically-induced cancer. An F2 generation of BDIV and BDIX rats [resistant and susceptible to ethylnitrosourea (ENU)-induced neuro-oncogenesis, respectively] was bred and subjected to ENU. Male hybrid rats developed malignant peripheral nerve sheath tumors twice as often as females and displayed a significantly shorter survival time. Six of seven Mss (mediating schwannoma susceptibility) loci influencing cancer risk showed sex-dependent effects. Homozygous BDIV alleles of the Mss4 locus on chromosome 6 were able to protect female hybrid rats almost exclusively from developing MPNST while 30% of males developed MPNST which corresponded to the average (BDIVxBDIX)F2 tumor rate [1]. Congenic BDIX animals which carried a 2,1 Mb fragment of the BDIV genome (97.6 - 99.7 Mb) confirmed the effect [2]. Mss4d includes the Esr2 gene known to mediate sex-specificity in a number of different traits.[3, 4] In parallel GWAS was performed using a F2 generation of BN and LE/Stm rats to elucidate the genetic architecture of thyroid neoplasia development induced by low iodine diet followed by i.v. application of methylnitrosourea (MNU). In this scenario sex-specificity was even stronger. 30% of males and 3% of females developed thyroid neoplasia. Three Mtns (mediating thyroid neoplasia susceptibility) loci were found to influence thyroid neoplasia risk in an allele–specific manner. Heterozygous or homozygous BN alleles at the Mtns1 locus on chromosome 6 appeared to mediate sex-specificity. This locus, too, includes the Esr2 gene. Therefore BDIV and BN alleles, only, appear to exert sex-specific effects while BDIX and LE/Stm alleles do not. To identify variant alleles DNA sequences of the Esr2 gene were compared using whole genome sequences generated by NGS from all four strains. Sequence variants shared by BDIV and BN rats in contrast to those shared by BDIX and LE/Stm rats appear to be a reasonable basis for unveiling the mechanism of Esr2-mediated sex dependence of traits.Daniel M. Gatti, Petr Simecek, Lisa Somes, Clifton T. Jeffrey, Matthew J. Vincent, Kwangbom Choi, Xingyao Chen, Gary A. Churchill, Karen L. Svenson
The Jackson Laboratory, 600 Main St, Bar Harbor, ME, 04609, USA
The Effects of Sex and Diet on Physiology and Liver Gene Expression in Diversity Outbred Mice
Inter-individual variation in adiposity and metabolic health is driven by many factors. Sex, genetic background, and diet, and the interactions between these factors, are significant determinants of propensity toward disease. How these factors affect variation in the expression of genes is also of critical importance in understanding the etiology of metabolic disorders. In this study, we fed 850 Diversity Outbred mice, half females and half males, either a standard chow diet or a high fat, high sucrose diet from wean age to 26 weeks. We measured clinical chemistries and body composition at early and late time points during the study, and liver transcription at euthanasia. Males weighed more than females and mice on the high fat diet generally weighed more than those on chow. Many traits showed sex- or diet-specific changes as well as more complex sex-by-diet interactions. We mapped both the physiological and transcript abundance traits and found that the genetic architecture of the physiological traits is complex, with many single locus associations being potentially driven by more than one polymorphism. Using an additive model, as well as models considering diet or sex effects, we found 28 loci for metabolic traits. For liver transcription, we found that local polymorphisms affect constitutive and sex-specific transcription, but that response to diet was not affected by local polymorphisms. We performed mediation analysis by mapping the physiological traits, given liver transcript abundance, and propose several genes that may be modifiers of the physiological traits. By including both physiological and gene expression traits in our analyses, we have created deeper outcome profiles to identify additional significant contributors to complex metabolic traits. We make the phenotype, liver transcript and genotype data publicly available as a resource for the research community.Danny Arends1, Sebastian Heise1, Stefan Kärst1, Gudrun A. Brockmann1
1Humboldt-Universität zu Berlin, Invalidenstraße 42, D-10115 Berlin, Germany
Fine mapping of a major obesity locus (jObes1)
The Berlin Fat Mouse Inbred line BFMI860 is a model system to study juvenile obesity. In previous research a recessive major effect locus (jObes1) was found to be responsible for 39% of the variance of total fat mass at 10 weeks in a (BFMI860 x C57BL/6NCrl) F2 population on chromosome 3 from 34 to 44 Mb.
To reduce this large interval an advanced intercross line (AIL) was generated from the initial F2 mapping population. Males of generation 28 were excessively phenotyped and genotyped using the MegaMuga mouse SNP chip. Expression of candidate genes was investigated in gonadal adipose tissue, liver and whole brain from mice of different genotype classes. Classical genetic complementation tests were performed to test the phenotype complementation ability of candidate genes.
The high mapping resolution of the AIL, combined with the high density of SNPs present on the MegaMuga array reduced the confidence interval for jObes1 from 10 Mb to 0.37 Mb between 36.48 and 36.85 Mb.
This region was highly significantly (LOD(BH) > 50) associated with total fat mass from puberty around 6 weeks on. Male homozygous carriers of the BFMI allele on the jObese1 locus had 3 grams more fat compared to the other genotypes. Surprisingly, this genotype class showed lower body mass until weaning at three weeks (LOD(BH) = 3.2). Using an AIL and the high density MegaMuga array the confidence interval for jObes1 could be reduced 27 fold by identifying chromosomal recombinations in the AIL population.
The reduced confidence interval contains four genes. Bbs7, deemed the most likely candidate gene since it also caused obesity in the complementation test and this gene was differentially expressed in all three examined tissues, while the neighboring gene Ccna2 only showed differential expression in gonadal adipose tissue.
Although Bbs7 is the most likely obesity gene in the jObes1 region, neighboring genes cannot be entirely excluded. Further examinations are needed to elucidate the molecular mechanism leading to physiological consequences on body mass and fat mass in juvenile animals.
Pjotr Prins1,6, Zachary Sloan1, Danny Arends2, Karl W. Broman3, Arthur Centeno1, Nicholas Furlotte8, Harm Nijveen4, Lei Yan1, Xiang Zhou5, Robert W. Williams1, Saunak Sen7
1University of Tennessee Health Science Center Department of Genetics, Genomics and Informatics, Memphis, TN 38163, USA
2Humboldt University, Berlin, Germany
3University of Wisconsin, USA
4Wageningen University, The Netherlands
5University of Michigan
6University Medical Center Utrecht, The Netherlands
7Department of Preventive Medicine, University of Tennessee Health Science Center, Memphis, TN 38163, USA
823andme, Inc.
GeneNetwork for web-based genetic analysis
GeneNetwork (GN) is a free and open source (FOSS) tool for web-based genetic analysis. GN allows biologists to upload high-throughput experimental data, such as expression data from microarrays and RNA-seq, and also `classic' phenotypes, such as disease phenotypes. These phenotypes can be mapped interactively against genotypes using embedded tools, such as R/QTL (Arends et al. 2010) mapping, interval mapping for model organisms and an implementation of FaST-LMM (Lippert et al. 2011) which is suitable for human populations and outbred crosses, such as the mouse diversity outcross. Interactive D3 graphics are included from R/qtlcharts and presentation-ready figures can be generated. Recently we have added functionality for phenotype correlation (Wang et al. 2016), network analysis (Langfelder and Horvath 2008) and Correlation Trait Loci (Arends et al., 2016). We are improving data upload mechanisms, meta-tagging, data integration and PheWAS-style analysis, as well as prediction methods and data query mechanisms, thereby improving GN as a resource for the mouse and rat research communities. In this talk I will present our current thoughts on where we want to go and how we want to achieve all this.